





Suivi de la gravité de la myasthénie au fil du temps : aperçu de la base de données des demandes d’indemnisation de l’assurance maladie française
Tracking myasthenia gravis severity over time: Insights from the French health insurance claims database
Actualité commentée réalisée par Guilhem Solé - Publiée le 22 janvier 2025
TKMR-N 2025 ; 4 (1) : A1
Attarian S, Camdessanché JP, Echaniz-Laguna A, Ciumas M, Blein C, Grenier B, et al.
Eur J Neurol 2025 ; 32 : e16518.
AVIS D’EXPERT
Cette étude longitudinale apporte une contribution à la compréhension de l’évolution de la myasthénie auto-immune (MA) en vie réelle, en exploitant une base de données nationale robuste (SNDS) couvrant plus de 99 % de la population française. L’échantillon de 14 459 patients est important et l’utilisation de critères bien définis pour identifier les cas assure une certaine fiabilité. Les résultats, notamment l’admission en soins intensifs et l’utilisation de traitements de secours pour certains patients, mettent en lumière le défi de contrôler efficacement la maladie chez un certain nombre de patients. L’analyse des facteurs de mortalité, notamment l’effet de l’âge, du sexe et des comorbidités, est cohérente avec la littérature existante.
Cependant, l’étude présente certaines limites. Tout d’abord, l’utilisation de données administratives, bien qu’avantageuse pour couvrir une large population, peut entraîner des biais de sélection. Par exemple, les patients présentant des formes légères de MA, qui ne nécessitent pas d’hospitalisation ou de déclaration en affection de longue durée (ALD), pourraient être sous-représentés. De plus, la base SNDS ne fournit pas d’information clinique détaillée, comme les résultats des tests ou les raisons précises des hospitalisations, ce qui limite la granularité des analyses. Par ailleurs, la dépendance à des modèles statistiques tels que Kaplan-Meier et Cox pourrait omettre des facteurs non mesurés influençant la gravité de la MA, comme l’accès aux centres spécialisés ou l’observance thérapeutique. La variation géographique des prévalences, attribuée à un potentiel sous-diagnostic dans les départements d’outre-mer, mérite une analyse plus approfondie pour valider cette hypothèse.
En conclusion, cette étude met en évidence les besoins médicaux non satisfaits dans la gestion de la MA, en particulier pour les patients gravement atteints.
Le nifuroxazide supprime les effets délétères associés aux défauts du MICOS associés à la CHCHD10 dans des modèles de maladie neurodégénératives
Nifuroxazide rescues the deleterious effects associated with CHCHD10-associated MICOS defects in disease models
Actualité commentée réalisée par Véronique Paquis-Flucklinger - Publiée le 03 décembre 2024
TKMR-N 2024 ; 3 (12) : A12
Ropert B, Bannwarth S, Genin EC, Vaillant-Beuchot L, Lacas-Gervais S, Madji Hounoum B, et al.
Brain 2024 : awae348.
AVIS D’EXPERT
Dans des maladies graves et incurables, telles que la SLA et les maladies mitochondriales, le repositionnement, qui consiste à utiliser un médicament dans une nouvelle indication, est considéré comme particulièrement intéressant. Le processus de repositionnement s’avère, en effet, plus rapide et moins coûteux que le développement de nouveaux médicaments. Comprendre les mécanismes à l’origine d’une pathologie est fondamental pour l’élaboration d’une stratégie thérapeutique. Le désassemblage du complexe MICOS ayant été identifié comme un élément clé dans les défauts provoqués par l’expression de la mutation p.S59L, les auteurs ont généré des levures mimant ce désassemblage. Les souches mutantes présentent un défaut de croissance qui a été utilisé pour tester plus de 1 600 médicaments quant à leur capacité à restaurer cette croissance. Parmi les deux composés identifiés, le nifuroxazide a été sélectionné pour une étude plus approfondie en raison de son effet important sur la croissance et, de sa faible toxicité. Cet antibiotique est utilisé en clinique par voie orale dans les diarrhées bactériennes. La découverte de l’inhibition de STAT3 par cette molécule avait déjà fortement soutenu son potentiel de repositionnement dans le traitement des cancers et des maladies inflammatoires.
L’étape suivante a été de tester les effets du nifuroxazide sur les défauts observés dans les cellules de patients. Les résultats montrent ses capacités à les corriger, y compris dans les modèles neuronaux. Pour mieux comprendre le mécanisme d’action du nifuroxazide, les auteurs ont également cherché à identifier ses cibles cellulaires. L’une d’entre elles semble être la protéine KIF5B, impliquée dans le transport des mitochondries le long des axones. Le nifuroxazide augmente le transport mitochondrial en dégradant la syntaphiline, une protéine qui arrête ce transport. Cet effet passe possiblement par la perturbation de l’interaction KIF5B/syntaphiline. D’autres études avaient proposé la syntaphiline comme une cible pharmacologique potentielle pour le traitement des maladies neurodégénératives. Ce travail a permis d’identifier le nifuroxazide comme une molécule capable de la dégrader.
La dernière étape consistera à tester le nifuroxazide in vivo sur un modèle de souris knock-in (Chchd10S59L/+) généré par les auteurs. Ces souris présentent tous les symptômes observés chez les patients et notamment des signes SLA typique avec dégénérescence des jonctions neuromusculaires, perte des motoneurones et agrégats TDP-43.
En conclusion, cette étude a permis d’identifier le nifuroxazide comme une molécule thérapeutique potentielle pour les troubles associés au désassemblage du MICOS et peut-être aussi pour certaines maladies neurodégénératives en raison de ses effets sur le transport mitochondrial. La réalisation d’études précliniques sur le modèle de souris Chchd10S59L/+ sera cruciale pour déterminer si ce médicament peut avoir un effet thérapeutique sur la neurodégénérescence observée chez ces animaux avant d’envisager un essai clinique chez l’homme.
Maladie ou imitation de maladie mitochondriale primaire : données d’une large cohorte française
Primary mitochondrial disorders and mimics: Insights from a large French cohort
Actualité commentée réalisée par Cécile Rouzier - Publiée le 26 juin 2024
TKMR-N 2024 ; 3 (11) : A11
Rouzier C, Pion E, Chaussenot A, Bris C, Ait-El-Mkadem Saadi S, Desquiret-Dumas V, et al.
Ann Clin Transl Neurol 2024 ; 11 : 1478-91.
AVIS D’EXPERT
Les variants dans les gènes nucléaires représentent jusqu’à 50 % des maladies mitochondriales primaires (PMD) dans certaines cohortes. Le réseau national français MITODIAG et les 2 centres de référence Maladies Mitochondriales CALISSON et CARAMMEL ont rapporté 397 patients français suspects de maladie mitochondriale chez qui un diagnostic moléculaire a pu être confirmé par la mise en évidence de variants pathogènes nucléaires par séquençage haut débit par panel, étude d’exome ou de génome. Cette étude montre la complexité à établir un diagnostic clinique de maladie mitochondriale. En effet, environ la moitié des patients (54 % [42/78] chez les enfants et 48 % [13/27] chez les adultes), ayant eu un séquençage d’exome (WES) ou de génome (WGS), présentaient des phénocopies, c’est-à-dire des pathologies non mitochondriales qui ressemblaient cliniquement aux maladies mitochondriales. Parmi ces patients, plus de la moitié présentait un déficit de la chaine respiratoire.
Une grande hétérogénéité clinique et génétique avec des variants identifiés dans 172 gènes différents a été observée. Chez les enfants avec PMD, une forte proportion de gènes étaient liés aux fonctions OXPHOS (sous-unités de la chaîne respiratoire, facteurs d’assemblage et cofacteurs), en particulier les sous-unités du complexe I, ainsi que dans les gènes impliqués dans la synthèse, la traduction et la dégradation des protéines mitochondriales, avec un pourcentage significatif de variants dans les gènes codant pour les synthétases et transférases de l’ARNt. Le syndrome de Leigh est la pathologie la plus fréquente. Chez les adultes, les variants pathogènes impliquent principalement des gènes de maintenance de l’ADNmt.
POLG est le gène le plus muté (28/397), et les phénotypes forment plutôt un continuum que des phénotypes distincts comme précédemment suggéré. Les enfants présentent une combinaison de différents symptômes, principalement épilepsie et hépatopathie, tandis que les adultes présentent principalement CPEO, ataxie et neuropathie. Rarement décrits, deux patients présentaient également un parkinsonisme.
Chez les patients présentant des pathologies imitant les maladies mitochondriales (PMD mimics), nous avons également observé une grande hétérogénéité génétique. Mis à part le gène SLC19A3, qui constitue le principal diagnostic différentiel du syndrome de Leigh « mitochondrial », aucun gène récurrent n’a été identifié. Les principales fonctions affectées incluent les facteurs de transcription/traduction, les canaux ioniques, les transporteurs, ou des protéines responsables de myopathies telles que la laminine, la titine ou la myosine.
Les approches pangénomiques de type WES ou WGS, plutôt que l’approche par panel, sont plus efficaces pour identifier un diagnostic moléculaire chez les patients avec des « possibles » pathologies.
Les auteurs ont proposé un arbre décisionnel pour guider les médecins dans leur stratégie diagnostique. Ils suggèrent de réaliser en premier lieu un séquençage de l’ADNmt, suivi d’un panel ciblé lorsque le score PMD est ≥ 5 (PMD probable). En l’absence de muscle disponible, le séquençage de l’ADNmt devrait être réalisé de préférence à partir de cellules uroépithéliales, qui sont plus sensibles à la détection des variants de l’ADNmt que le sang. En cas de consanguinité, le séquençage des gènes nucléaires devrait être réalisé en premier lieu. La biopsie musculaire n’est plus utilisée en routine comme diagnostic de première intention, mais elle reste essentielle pour le diagnostic génétique précis des grandes délétions de l’ADNmt, notamment pour les CPEO, et des variants de l’ADNmt restreints au muscle squelettique. La biopsie musculaire est également recommandée en cas de maladie grave nécessitant un diagnostic rapide. De plus, compte tenu de l’augmentation significative du nombre de variants de signification inconnue (VUS) identifiés, la biopsie musculaire et/ou les biopsies cutanées sont essentielles pour réaliser des études fonctionnelles afin de soutenir la pathogénicité du variant. Enfin, il peut également être utile de trouver des arguments supplémentaires pour le diagnostic de PMD lorsque la génétique est non concluante.
L’inhibition de RANKL réduit l’hypertrophie cardiaque chez les souris mdx et possiblement chez les enfants avec dystrophie musculaire de Duchenne
RANKL Inhibition Reduces Cardiac Hypertrophy in mdx Mice and Possibly in Children with Duchenne Muscular Dystrophy
Actualité commentée réalisée par Catherine Sarret - Publiée le 14 juin 2024
TKMR-N 2024 ; 3 (10) : A10
Marcadet L, Juracic ES, Khan N, Bouredji Z, Yagita H, Ward LM, et al.
Cells 2023 ; 12 : 1538.
AVIS D’EXPERT
Le zolédronate (biphosphonate) est une molécule aujourd’hui bien établie, utilisée en perfusion intraveineuse semestrielle sur plusieurs heures dans la prise en charge de l’ostéopénie des patients DMD. Il est toutefois source de potentielle mauvaise tolérance avec induction de syndromes post-grippaux. En revanche, le dénosumab, anticorps monoclonal anti-RANKL, qui constitue une nouvelle thérapie dans la prise en charge de l’ostéoporose chez la femme adulte, ne dispose pas d’indication dans les pathologies neuromusculaires de l’enfant. Son utilisation commence à se développer dans les pathologies osseuses de l’enfant. Les données précliniques et cliniques de cette étude et les précédentes publications sur l’impact de la voie RANKL/RANK sur le muscle squelettique sont encourageantes dans la DMD avec un double intérêt osseux et musculo-cardiaque, ce d’autant qu’il n’existe pas d’effet indésirable cardiaque décrit et que l’injection sous-cutanée rapide semble mieux tolérée que la perfusion de zolédronate. En revanche des hypercalcémies rebonds ont été décrites en particulier à l’arrêt des traitements chez les enfants porteurs de pathologies osseuses et sont à surveiller1,2. Des études cliniques plus élargies sont attendues dans les pathologies neuromusculaires.
La déficience en SNUPN provoque une dystrophie musculaire récessive due à une altération de l’épissage des ARN et à une dysrégulation de la matrice extracellulaire
SNUPN deficiency causes a recessive muscular dystrophy due to RNA mis-splicing and ECM dysregulation
Actualité commentée réalisée par Fernández Gorka - Publiée le 15 avril 2024
TKMR-N 2024 ; 3 (9) : A9
Nashabat M, Nabavizadeh N, Saraçoğlu HP, Sarıbaş B, Avcı Ş, Börklü E, et al.
Nat Commun 2024 ; 15 : 1758.
AVIS D’EXPERT
Les auteurs décrivent une nouvelle forme de dystrophie musculaire infantile reliée à des variants récessifs du gène SNUPN. L’atteinte multisystémique, associant volontiers des cataractes et une atteinte du système nerveux central, est observée dans d’autres dystrophies musculaires avec altération du splicéosome, telle que la maladie de Steinert. Les auteurs approfondissent le mécanisme pathophysiologique de la maladie en démontrant un défaut d’oligomérisation de la protéine SPN1. En outre, les analyses transcriptomiques révèlent un dérèglement de l’épissage et de l’expression des ARNm entraînant une perturbation de l’organisation du cytosquelette, reliant ainsi cette nouvelle maladie à d’autres dystrophies musculaires congénitales impliquant les composants de la matrice extracellulaire, telles que la myopathie reliée au gène LAMA2 et les alpha-dystroglycanopathies.
Des variants bialléliques de SNUPN provoquent une dystrophie musculaire des ceintures avec des caractéristiques de type myofibrillaire
Biallelic variants in SNUPN cause limb girdle muscular dystrophy with myofibrillar-like features
Actualité commentée réalisée par Tanya Stojkovic - Publiée le 29 mars 2024
TKMR-N 2024 ; 3 (8) : A8
Iruzubieta P, Damborenea A, Ioghen M, Bajew S, Fernandez-Torron R, Töpf A, et al.
Brain 2024 : awae046. [Epub ahead of print].
AVIS D’EXPERT
Les auteurs de cet article ont réalisé un travail conséquent pour démontrer la fonction de ce gène et son action finalement sur la machinerie d’épissage, capitale pour la survie des cellules.
L’originalité de cet article est de démontrer qu’il existe une régulation très fine de la maturation des ARN messagers avec des conséquences sur l’expression de nombreuses protéines. Toutefois, le phénotype des patient mutés dans cet article pour SNUPN reste restreint au muscle strié. Néanmoins, d’autres mutations tronquantes de SNUPN identifiées récemment1 révèlent que l’atteinte peut être multisystémique, associant volontiers une myopathie des ceintures, une cataracte, une atteinte du système nerveux central et une cardiopathie, comme attendu étant donné son rôle important dans le splicéosome. Ceci est en concordance avec la physiopathologie connue pour d’autres myopathies ayant un impact sur le splicéosome, telle que la maladie de Steinert.
Le modèle drosophile montre que le gène SNUPN invalidé montre une réduction de mobilité et de viabilité des mouches mais il aurait été intéressant de créer un modèle knock-in avec la mutation identifiée chez les patients afin de mieux comprendre la physiopathologie de cette myopathie et les dérégulations sur le complexe ribonucléique.
Il reste à comprendre le lien entre snuportine-1 et formation des agrégats sarcoplasmiques protéiques ainsi que les mécanismes qui la lient à l’activation de l’autophagie.
Quantification de l’atteinte musculaire par IRM dans la polyneuropathie amyloïde familiale
Quantification of muscle involvement by MRI in familial amyloid polyneuropathy
Actualité commentée réalisée par Shahram Attarian - Publiée le 12 mars 2024
TKMR-N 2024 ; 3 (7) : A7
Durelle C, Delmont E, Michel C, Trabelsi A, Hostin MA, Ogier A, et al.
Eur J Neurol 2023 ; 30 : 3286-95.
AVIS D’EXPERT
Sur la base de mesures d’IRM quantitatives combinées à une méthode de segmentation dédiée, nous avons rapporté des altérations musculaires au niveau de la cuisse et de la jambe chez 39 patients porteur d’une mutation de la transthyrétine. De plus, ces mesures IRM ont été corrélées à plusieurs scores cliniques et mesures électrophysiologiques. Ces observations suggèrent que l’IRM musculaire quantitative pourrait constituer un biomarqueur intéressant dans le suivi de la polyneuropathie amyloïde familiale. Le MTR était diminué et la FF était augmentée pour plusieurs muscles du groupe symptomatique avec une atteinte préférentielle des compartiments postérieurs et latéraux. Ces résultats suggèrent que le phénotype d’atteinte de la TTR-FAP affectent principalement les compartiments musculaires postérieurs et notamment le gastrocnémius latéralis. De plus, dans le groupe asymptomatique, les mesures d’IRM quantitative étaient presque inchangées à l’exception d’une augmentation de la FF dans ce même muscle, suggérant qu’il pourrait s’agir du premier muscle atteint dans la polyneuropathie amyloïde familiale. La surveillance spécifique de ce muscle par IRM pourrait être un bon indicateur du passage d’une forme asymptomatique vers une forme symptomatique de la maladie. Trois muscles (le sartorius, le gracilis et le gastus médialis) présentaient un MTR diminué mais une FF dans les normes. Pour ces muscles particuliers, le MTR pourrait être plus sensible à des changements subtils du contenu macromoléculaire, survenant avant le début du processus de FF. En d’autres termes, le MTR pourrait être considéré comme un marqueur précoce du processus neuropathique. Un suivi de ces biomarqueurs serait intéressant afin d’évaluer leur évolution, notamment en réponse au traitement.
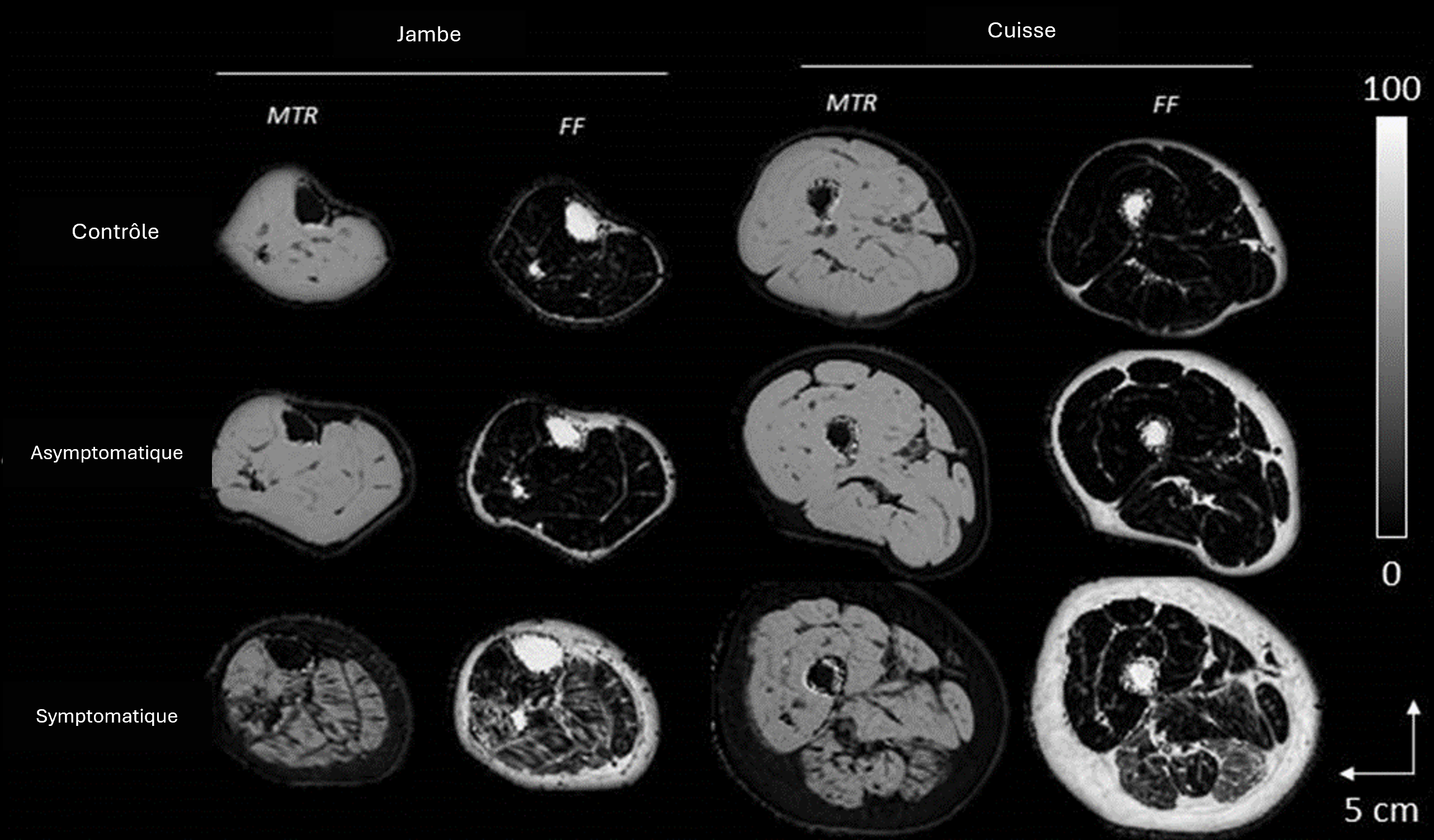
Abréviations : FF : infiltration graisseuse ; MTR : transfert d’aimantation.
Figure 1 : Cartes de l’infiltration graisseuse et du transfert d’aimantation chez un sujet contrôle, asymptomatique et symptomatique (PND 3, NIS-LL 40 et 1 an d’évolution de la maladie).
On observe une atteinte préférentiellement postérieure et latérale chez le sujet symptomatique.
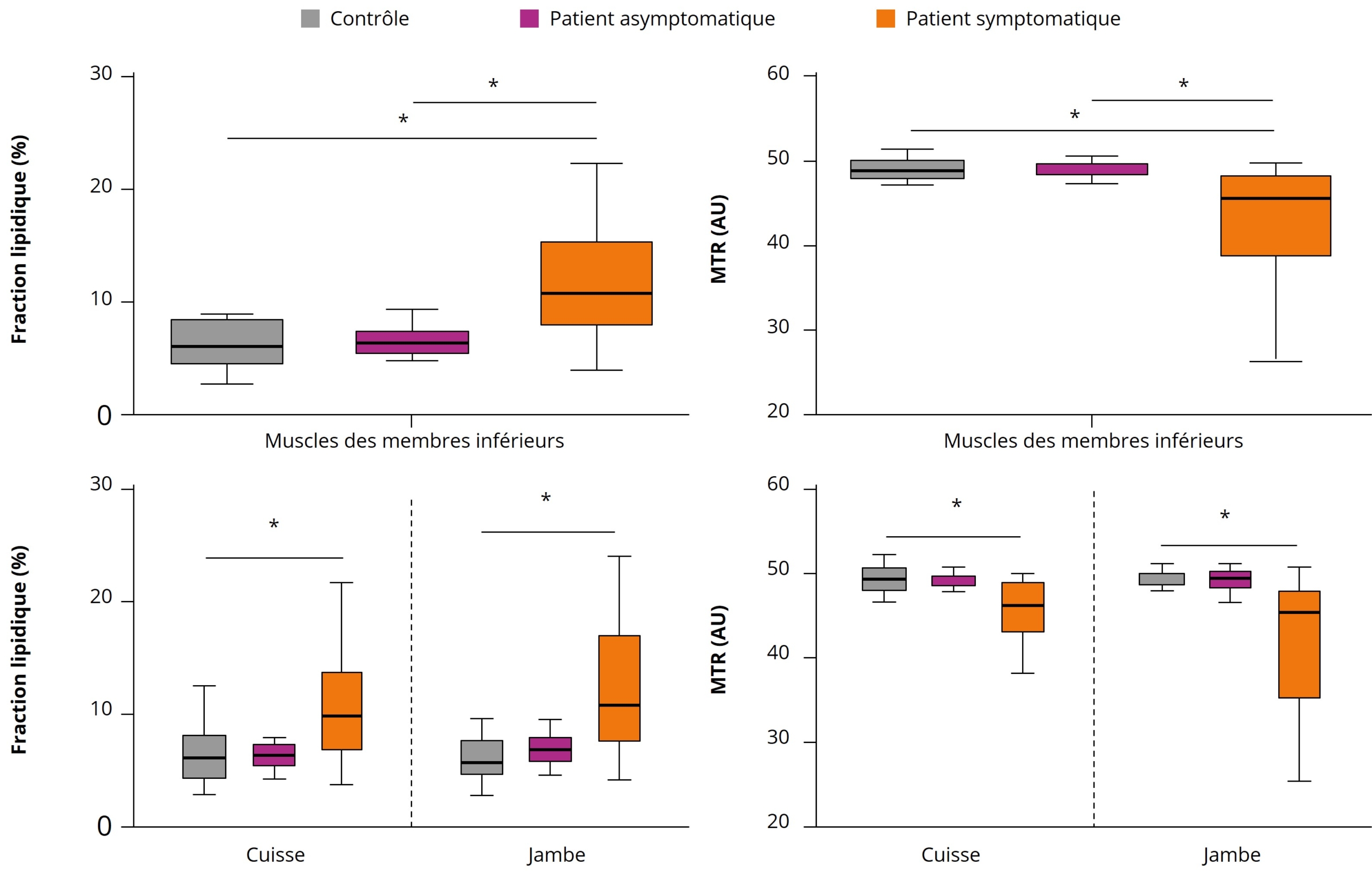
*indique des valeurs de p < 0,05
Figure 2 : Infiltration graisseuse (%) et transfert d’aimantation (AU) global du membre inférieur chez les sujets contrôles, asymptomatiques et symptomatiques.
La FF était significativement augmentée et le MTR significativement diminué chez les sujets symptomatiques. Il n’y avait pas de différence entre les sujets asymptomatiques et les contrôles.
Effet du traitement par nusinersen à 3 ans de traitement chez 57 amyotrophies spinales corrélé au nombre de copies SMN2 et du type initial
Effect of nusinersen after 3 years of treatment in 57 young children with SMA in terms of SMN2 copy number or type
Actualité commentée réalisée par Isabelle Desguerre - Publiée le 05 mars 2024
TKMR-N 2024 ; 3 (6) : A6
Audic F, Dubois SM, Durigneux J, Barnerias C, Isapof A, Nougues MC, et al.
Arch Pediatr 2023 : S0929-693X(23)00211-7. [Epub ahead of print]
AVIS D’EXPERT
Cet article rapporte l’expérience de la filière française FILNEMUS des maladies neuromusculaires en vie réelle pour les 57 premiers enfants ASI type 1 et 2 naïfs dont le traitement a été initié entre 2017 et 2019 avec un recul de 3 ans. L’homogénéité de la prise en charge et du suivi sur tout le territoire français permet de mieux rendre compte du bénéfice de ce traitement initié précocement sur le plan moteur, respiratoire et déglutition en particulier chez les enfants porteurs de 3 copies SMN2. Ces résultats confortent sur des séries plus petites et moins homogènes le rôle du nombre de copies SMN2 sur le pronostic spontané mais aussi sur la qualité de la réponse au traitement comme suggéré dans la littérature. Les résultats de cette série sont plus favorables que les autres études mais confirment la gravité pronostic des types 1a/b avec 2 copies SMN2 malgré un traitement initié le plus rapidement possible après le diagnostic. Il confirme aussi l’absence de réversibilité de l’atteinte respiratoire et bulbaire et de la nécessité de support respiratoire ou nutritionnel chez les enfants type 1a/b. La scoliose précoce reste un problème majeur chez les patients porteur de 2 copies mais aussi pour plus de 50 % des 3 copies malgré une bonne réponse motrice. La possibilité de scolarisation dès l’âge de 3 ans et demi est facilitée chez les enfants autonomes sur le plan respiratoire et nutritionnel.
L’atteinte des muscles faciaux est fréquente dans la myosite à inclusions
Face to Face: deciphering facial involvement in inclusion body myositis
Actualité commentée réalisée par Emmanuelle Salort-Campana - Publiée le 01 mars 2024
TKMR-N 2024 ; 3 (5) : A5
Fortanier E, Delmont E, Kouton L, Corazza G, Grapperon AM, Verschueren A, et al.
J Neurol 2024 ; 271 : 410-8.
AVIS D’EXPERT
La myosite à inclusions est une affection musculaire relativement fréquente, historiquement classée au sein des myopathies inflammatoires bien qu’elle se distingue très clairement des autres myosites, en particulier par son absence de réponse aux traitements immunosuppresseurs classiques. Le diagnostic peut parfois être délicat car il n’y a pas de marqueur sérologique et les critères anatomopathologiques peuvent être mis en défaut, notamment au début de la maladie. La présence d’une faiblesse des muscles faciaux dans les myopathies est rare et en règle générale absente dans le groupe des myopathies inflammatoires. L’atteinte faciale dans l’IBM, classiquement décrite, a donc une valeur discriminante pour distinguer cette entité des autres myosites. Cependant, à ce jour, aucune étude prospective n’a étudié formellement la prévalence et les caractéristiques de celle-ci. Il faut souligner la difficulté de l’étude de l’atteinte faciale dans les myopathies étant donné qu’il n’existe pas de méthode validée, ni de gold standard.
L’étude de Fortanier et al. a exploré l’atteinte faciale chez les patients IBM en utilisant un SAF précédemment utilisé dans une étude hollandaise portant sur des patients atteints de FSHD (Loonen et al., 2021). La présence ou non d’une atteinte faciale était déterminée par des valeurs du SAF se situant au-dessus de celles d’un groupe de contrôles sains. Pour permettre une bonne validité des résultats, les vidéos ont été cotées par 5 examinateurs différents, de manière indépendante, avec une bonne reproductibilité inter et intra évaluateur.
Cette étude a permis d’estimer la fréquence de l’atteinte faciale dans l’IBM à plus de 50 % alors qu’elle était estimée à 87 % des patients FSHD comparativement à la limite supérieure des contrôles sains. Elle a montré que l’atteinte prédominait sur la partie supérieure de la face, notamment sur les orbiculaires des paupières. Les résultats suggèrent que les patients IBM avec une atteinte faciale ont une atteinte de la déglutition plus importante mais ceci nécessite pour confirmation une étude portant sur un plus grand nombre de patients.
L’atteinte faciale était plus sévère chez les patients FSHD que chez les IBM. Cette atteinte de la face plus subtile chez les patients IBM se reflétait également par une discordance inter observateur lorsque l’existence d’une atteinte faciale n’était basée que sur une évaluation qualitative. Ceci souligne l’intérêt de l’utilisation du SAF pour évaluer l’atteinte faciale dans l’IBM et possiblement de manière plus large dans les maladies musculaires.
Des précisions sur le rôle du microbiote intestinal dans la dystrophie musculaire de Duchenne : une étude longitudinale chez la souris mdx
Insight into the role of gut microbiota in Duchenne Muscular Dystrophy: an age-related study in mdx mice
Actualité commentée réalisée par Jon Andoni Urtizberea - Publiée le 05 février 2024
TKMR-N 2024 ; 3 (4) : A4
Jollet M, Mariadassou M, Rué O, Pessemesse L, Ollendorff V, Ramdani S, et al.
Am J Pathol 2023 ; S0002-9440.
AVIS D’EXPERT
Le terme de microbiote recouvre la population de plusieurs milliards de bactéries qui peuplent le tractus digestif dont notre intestin. Celles-ci participeraient de l’homéostasie de l’organisme et donc, in fine, de la santé. Des travaux récents ont révélé l’existence de communications fonctionnelles entre l’intestin et d’autres organes comme le cerveau, le cœur, le foie ou le tissu adipeux. Des dérèglements de cette flore intestinale seraient à l’origine d’interférences, d’ordre épigénétique, avec des pathologies dégénératives comme la maladie d’Alzheimer ou la dépression, ou avec des pathologies endocriniennes, dont le diabète et l’obésité. Des expérimentations récentes prônent même les vertus thérapeutiques, dans ces pathologies, des greffes de microbiote ou de l’administration orale d’agents probiotiques.
L’article cité en référence étudie, de manière très rationnelle et documentée, les liens entre microbiote et muscle squelettique, et plus particulièrement chez la souris modèle de la DMD. En ce sens, ces travaux constituent une première. Le microbiote de la souris mdx est profondément modifié et les études fonctionnelles plaident en faveur d’un axe muscle-microbiote, ce qui ne constitue qu’une demi-surprise si l’on considère que le muscle représente 40 à 50 % du poids d’un individu.
Un des rationnels de l’étude repose sur les travaux de Bindels et al1. Celui-ci avait étudié des souches de souris leucémiques traitées et présentant un phénotype cachectique et démontré qu’un traitement par probiotiques diminuait l’inflammation du muscle squelettique et l’amyotrophie. On sait également que les souris privées artificiellement de microbiote ont une masse musculaire moindre.
Ces études restent, selon nous, encore très spéculatives, et procèdent pour partie de l’engouement suscité par les travaux plus généraux sur le microbiote et son implication en pathologie humaine.
Par ailleurs, on connait les limites du modèle murin de la DMD : la souris mdx n’a pas un phénotype totalement superposable à celui de la maladie humaine. Il n’est pas exclu non plus que beaucoup de facteurs soient fortement intriqués. Les troubles du transit, très fréquents chez les patients atteints de DMD (surtout quand ils avancent en âge), favorisent la stase fécale et donc indirectement la flore intestinale. Les auteurs citent également le rôle perturbateur des antibiothérapies chez eux.
De là à imaginer une approche thérapeutique susceptible de changer l’histoire naturelle de la DMD, il y a un grand pas que certains n’hésitent pourtant pas à explorer2.
Un nouveau variant faux-sens G376V du gène codant la TDP-43 entraine une myopathie distale de début tardif et pas une sclérose latérale amyotrophique
The new missense G376V-TDP-43 variant induces late-onset distal myopathy but not amyotrophic lateral sclerosis
Actualité commentée réalisée par Jon Andoni Urtizberea - Publiée le 01 février 2024
TKMR-N 2024 ; 3 (3) : A3
Zibold J, Lessard LER, Picard F, da Silva LG, Zadorozhna Y, Streichenberger N, et al.
Brain 2023 : awad410. Epub ahead of print.
AVIS D’EXPERT
Le papier cité en référence confirme l’implication de TDP-43 dans une myopathie distale. Les études fonctionnelles sont, de ce point de vue, très convaincantes. L’accumulation de la protéine au sein du muscle mais aussi à l’intérieur de vacuoles apparues dans des myoblastes de patients sont des éléments de preuve indiscutables.
Le variant G376V, bien que faux-sens et donc d’interprétation plus délicate que d’habitude, était inconnu des bases de données relatives aux myopathies distales. La ségrégation au sein des deux familles allait également dans le sens de la pathogénicité. Le fait qu’il s’agisse du même variant dans deux familles géographiquement proches laisse planer un doute en faveur d’un effet fondateur.
L’originalité de cette observation tient dans le caractère exclusivement myopathique du phénotype. C’est sans doute ce caractère inhabituel qui a retardé l’élucidation, au niveau moléculaire, de ces deux familles bien connues pour avoir faire l’objet de plusieurs communications au sein de la communauté myologique française. Il y a plus de 15 ans, et en l’absence de séquençage à haut débit, les premières études génétiques s’étaient en effet concentrées sur les gènes connus pour être responsables de myopathie distale. Parmi ces derniers, le gène MATR3 codant la matrine de type 3, un moment envisagé, car incriminé lui-aussi, dans des phénotypes mixtes de type SLA ou de type myopathique s’est avéré être une fausse piste.
Les auteurs estiment, à juste raison, qu’il faut désormais incorporer ce gène (TARDBP) dans le panel des gènes de myopathie distale même s’il n’avait été, jusqu’ici, impliqué que dans des formes familiales de SLA.
De manière plus générale, plusieurs gènes, comme VCP ou MATR3 ont déjà été impliqués dans des phénotypes dits de chevauchement entre pathologie motoneuronale et myopathie. TARDPBP vient donc de rejoindre cette liste appelée à s’étendre.
Une technologie optique de cartographie génomique dans l’aide au diagnostic moléculaire des patients suspects de dystrophie musculaire facio-scapulo-humérale
Molecular diagnosis of facioscapulohumeral muscular dystrophy in patients clinically suspected of FSHD using optical genome mapping
Actualité commentée réalisée par Jon Andoni Urtizberea - Publiée le 17 janvier 2024
TKMR-N 2024 ; 3 (2) : A2
Guruju NM, Jump V, Lemmers R, Van Der Maarel S, Liu R, Nallamilli BR, et al.
Neurol Genet 2023 ; 9 : e200107.
AVIS D’EXPERT
Le diagnostic moléculaire de la FSHD reste complexe car la physiopathologie de cette maladie, déroutante à bien des égards, l’est tout autant. Cette pathologie reste hétérogène quant aux mécanismes et réarrangements chromosomiques impliqués. Les techniques de biologie moléculaire jusqu’ici utilisées traditionnellement étaient particulièrement adaptées aux formes classiques de la forme la plus commune de myopathie FSHD1. Pour autant, elles ne restent maitrisées que par un tout petit nombre de laboratoires dans le monde et n’ont pu bénéficier de la révolution en cours en matière de séquençage à haut débit (NGS pour next-generation sequencing).
L’approche très intégrative proposée par ce nouveau test à visée diagnostique est donc très encourageante et bienvenue à l’heure où un très grand nombre d’authentiques malades atteints de FSHD n’ont pas, à travers le monde, de diagnostic de confirmation. Les résultats de validation obtenus dans l’article cité en référence sont très honorables et le rendement diagnostique tout à fait comparable à d’autres méthodes. On peut imaginer que le test sera, dans sa version commerciale, moins chronophage et possiblement, l’avenir nous le dira, moins onéreux.
L’originalité de l’OGM tient surtout dans sa capacité à interpréter de manière automatique, après une phase d’apprentissage, les réarrangements observés et les haplotypes associés. En la couplant avec l’étude du gène de la FSHD2 par NGS, on obtient ainsi, et à grande échelle, une cartographie fine des génotypes de la FSHD, y compris les plus complexes d’entre eux.
On notera enfin qu’une approche similaire avait déjà été utilisée par le laboratoire de génétique de Marseille (peignage moléculaire) et que l’OGM est en concurrence directe avec un autre laboratoire, Oxford Nanopore, qui explore les capacités d’un outil plus fiable et plus rapide pour le diagnostic moléculaire de la FSHD.
Une relation dose-effet lors de la prise d’hydrates de carbone avant l’exercice en situation d’indisponibilité glycogénique : les leçons de la maladie de McArdle
Dose-response effect of pre-exercise carbohydrates under muscle glycogen unavailability: Insights from McArdle disease
Actualité commentée réalisée par Jon Andoni Urtizberea - Publiée le 04 janvier 2024
TKMR-N 2024 ; 3 (1) : A1
Valenzuela PL, Santalla A, Alejo LB, Merlo A, Gustos A, Castellote-Bellés L, et al.
J Sport Health Sci 2023 : S2095-546.
AVIS D’EXPERT
La maladie de McArdle intéresse beaucoup les spécialistes du sport en général et ceux de la physiologie musculaire en particulier. Elle représente en effet l’archétype de l’intolérance à l’effort en lien avec une indisponibilité fonctionnelle en glycogène. On en veut pour preuve le fait que l’article cité en référence émane d’une unité de recherche espagnole spécialisée dans le domaine du sport. La prise en charge et la préparation, au sens large, des sportifs, pourrait donc tirer bénéfice de ces travaux réalisés chez le sujet malade.
La principale originalité de cette étude réside dans le dosage très élevé (150 g) de l’association glucose-fructose proposée comme bras dans le protocole. On notera également que le laps de temps entre l’ingestion du produit et le démarrage de l’exercice était beaucoup plus réduit (10-15) que dans celui proposé dans d’autres protocoles expérimentaux chez l’homme (40 minutes). Cette dose semble plus efficace que celle, plus classique, de 75 g. Pour autant, et comme le soulignent les auteurs, elle ne permet pas d’atteindre une récupération complète des capacités énergétiques et de l’endurance.
La méthodologie utilisée ici est acceptable même si elle concerne un nombre limité d’individus testés. A contrario, ceux-ci étaient phénotypiquement et génotypiquement homogènes selon l’échelle de sévérité développée pour le registre espagnol des patients atteints de maladie de McArdle. Les paramètres étudiés sont plus fournis que dans d’autres protocoles comparables. Enfin, l’étude sur modèles cellulaires est également intéressante bien que de portée plus limitée.
On regrettera enfin qu’il ne soit pas fait mention dans cet article du principal frein, en vie réelle, à ce type de recommandation, à savoir le risque de surcharge pondérale à long terme. En effet, 150 g correspondent grosso modo à 24 morceaux de sucre du commerce…
Le séquençage de l’exome entier fait ressortir une association entre variants rares des gènes CTCF, DNMT1, DNMT3A, EZH2 et SUV39H1 et myopathie FSHD
Whole exome sequencing highlights rare variants in CTCF, DNMT1, DNMT3A, EZH2 and SUV39H1 as associated with FSHD
Actualité commentée réalisée par Jon Andoni Urtizberea - Publiée le 15 novembre 2023
TKMR-N 2023 ; 2 (22) : A22
Strafella C, Caputo V, Bortolani S, Torchia E, Megalizzi D, Trastulli G, et al.
Front Genet 2023 ; 14 : 1235589.
AVIS D’EXPERT
La physiopathologie de la dystrophie musculaire de type FSHD reste, encore à ce jour, d’une grande complexité. Si la responsabilité de la protéine DUX4 semble bien établie depuis les travaux de l’équipe de Leiden aux Pays-Bas1, les facteurs permettant d’expliquer la variabilité phénotypique souvent observée au sein d’une même famille restent en grande partie inconnus. Il est très vraisemblable que plusieurs gènes modificateurs influent sur le phénotype. Un premier pas a été franchi ces dernières années avec la découverte de variants dans trois gènes : SMCHD1, LRIF1 et DNMT3B. Les auteurs italiens de l’article cité en référence ont fait l’hypothèse que d’autres variants pathologiques pourraient être en cause et ont, dans ce but, décider de tester un très grand nombre de gènes grâce à une approche de type WES. Pour autant, et c’est à peu près le seul biais présent dans cette étude, les chercheurs ont concentré leur attention sur une liste, assez impressionnante il est vrai, mais pas exhaustive, de gènes candidats du fait de leurs liens supposés avec la FSHD. Ont été logiquement ciblés des gènes en rapport avec l’organisation de la chromatine et en particulier la méthylation. Les auteurs soulignent toutefois l’absence de corrélation stricte, pour les cinq nouveaux gènes identifiés, entre la taille du fragment D4Z4, le niveau de méthylation et les signes cliniques, limitant ainsi la portée de tels travaux et appelant à une certaine prudence quant à l’interprétation des résultats.
En complément de l’identification de gènes modificateurs, il apparait que l’étude de la méthylation est une source importante, voire primordiale, de renseignements pour expliquer la variabilité phénotypique, le caractère complet ou incomplet de la pénétrance, et peut-être pour détecter les sujets présymptomatiques. En conclusion, ces travaux ont le grand mérite d’élargir la palette des gènes modificateurs dans la dystrophie musculaire facio-scapulo-humérale et de confirmer la grande complexité de sa physiopathologie.
Issue fatale chez un patient atteint de dystrophie musculaire de Duchenne et traité par une thérapie génique à haute dose médiée par un AAV-9 recombinant
Death after High-Dose rAAV9 Gene Therapy in a Patient with Duchenne’s Muscular Dystrophy
Actualité commentée réalisée par Jon Andoni Urtizberea - Publiée le 06 novembre 2023
TKMR-N 2023 ; 2 (21) : A21
Lek A, Wong B, Keeler A, Blackwood M, Ma K, Huang S, et al.
NEJM 2023 ; 389 : 1203-10.
AVIS D’EXPERT
Le premier essai de thérapie génique comprenant un outil d’édition génomique empaqueté dans un AAV-9 s’est donc achevé sur un échec. En amont de l’essai, de nombreuses discussions avaient eu lieu sur la pertinence et le timing de cette première expérimentation chez l’homme, certains la jugeant beaucoup trop prématurée. Le patient était certes à un stade avancé de sa maladie mais semblait en capacité, au vu des paramètres cardiaque et respiratoire de base, de supporter le stress que représente une charge virale aussi importante (1 x 1014 de génomes-vecteurs par kilogramme de poids constitue une dose élevée). Cela n’a visiblement pas été le cas comme l’a prouvé la survenue de ce décès précoce. Les auteurs invoquent plus l’immunité innée contre les capsides virales que les autres réactions immunitaires telles l’activation du complément. Ils n’ont pas retrouvé d’arguments formels en faveur d’une microangiopathie thrombotique, par exemple, bien que les plaquettes aient eu tendance à diminuer significativement en post-injection. D’après eux, le parallèle devrait plutôt être fait avec un autre patient lui aussi décédé très précocement, à J6 post-injection, d’un choc cardiogénique au cours d’un autre essai de thérapie génique AAV-9 à base de fordadistrogene movaparvovec. Comme toujours en la matière, il faut savoir apprendre de ses échecs. On a cru un moment que cet essai à l’issue tragique porterait un coup fatal au développement de l’édition génomique, ce d’autant que d’autres essais similaires, mais dans d’autres pathologies, ont connu aussi des problèmes de tolérance. Les éléments de preuve accumulés par les investigateurs permettent d’écarter la responsabilité directe du transgène CRISPR-Cas9 mais soulignent, a contrario, qu’un terrain trop débilité fait sans doute prendre trop de risques au malade atteint de DMD. Ceci donne des indications précieuses sur les obstacles à la thérapie génique DMD à un âge avancé.
Les associations de thérapies innovantes dans l’amyotrophie spinale : proposition d’une classification
Combination disease-modifying treatment in spinal muscular atrophy: A proposed classification
Actualité commentée réalisée par Jon Andoni Urtizberea - Publiée le 16 octobre 2023
TKMR-N 2023 ; 2 (20) : A20
Proud CM, Mercuri E, Finkel RS, Kirschner J, De Vivo DC, Muntoni F, et al.
Ann Clin Transl Neurol 2023 ; (Online, Epub ahead of print).
AVIS D’EXPERT
Les praticiens ayant à traiter les enfants atteints de SMA n’ont que l’embarras du choix entre un produit de thérapie génique (l’OA), des oligonucléotides antisens injectés par voie intrathécale (nusinersen), ou une petite molécule agissant sur l’épissage du gène SMN2 (risdiplam). Ceci est surtout vrai pour les plus jeunes des patients SMA (âgé de moins de deux ans) qui sont, pour l’instant, les seuls éligibles à l’administration d’OA. Faute d’études comparatives entre les trois produits, les thérapeutes prescrivent ces produits de manière empirique en monothérapie au gré des disponibilités locales, de l’expérience des prescripteurs, et des conditions de remboursement. Les choses deviennent encore plus complexes lorsqu’il s’agit de changer de thérapie ou, plus rarement, de les associer simultanément. Pour l’essentiel, ces changements sont motivés par la relative inefficacité ou l’épuisement du bénéfice d’un premier produit, et/ou par la survenue d’effets secondaires de gravité variable.
Dans ce contexte, l’étude citée en référence donne une vision d’ensemble des habitudes des prescripteurs et de la topologie des changements thérapeutiques communément appelés « add-ons », « switchs » ou « bridges », ces derniers correspondant, par exemple, à une thérapie d’attente si le produit cible n’est pas encore disponible. Ce recensement permet ainsi de classer les différents types de stratégie thérapeutique, de voir celles qui émergent et celles qui posent éventuellement problème.
De manière générale, le phénomène des bi- ou trithérapies est loin d’être négligeable car il concerne 43 % des patients à ce jour. Il n’est pour autant étayé par aucun rationnel scientifique précis et n’est pas, jusqu’à présent, remis en cause par les payeurs.
L’analyse, pour aussi pertinente qu’elle soit, n’est pas exempte de limitations. Avec 443 cas étudiés, le registre RESTORE ne représente qu’une toute petite partie de la vingtaine de milliers de patients SMA qui sont exposés à l’un ou à plusieurs de ces trois médicaments. Le fait qu’il soit sponsorisé et maintenu par le laboratoire induit vraisemblablement aussi un biais de recrutement au profit de l’OA.
Malgré cela, le consensus d’experts proposé ici concernant les termes à utiliser pour décrire les schémas thérapeutiques s’avèrera très utile dans la perspective d’analyses comparatives de ces produits.
Evaluation numérique des fonctions respiratoires et du membre supérieur dans l’amyotrophie spinale : à propos d’un ensemble d’outils développés sur un smartphone (design, faisabilité, fiabilité et validité)
Digital measures of respiratory and upper limb function in spinal muscular atrophy: design, feasibility, reliability, and preliminary validity of a smartphone sensor-based assessment suite
Actualité commentée réalisée par Jon Andoni Urtizberea - Publiée le 06 octobre 2023
TKMR-N 2023 ; 2 (19) : A19
Perumal TM, Wolf D , Berchtold D, Pointeau G, Zhang YP, Cheng WY, et al.
Neuromuscul Disord 2023 : (Online, Epub ahead of print).
AVIS D’EXPERT
Le suivi habituel des patients atteints de SMA, traités ou non par une molécule innovante, passe par des évaluations cliniques en milieu hospitalier à intervalles réguliers. La pandémie de COVID-19 a montré les limites et les lourdeurs d’une telle organisation et a donné une impulsion sans précédent au développement d’outils d’évaluation à distance.
L’application développée sur smartphone et testée par une partie des investigateurs de l’essai JEWELFISH vient à point nommé pour illustrer cette tendance.
La corrélation avec certains des paramètres utilisés en clinique s’est avérée très encourageante. Comme le souligne les auteurs, ce nouvel outil permet ainsi une évaluation certes partielle, mais plus rapide, plus régulière (entre deux visites de suivi à l’hôpital) et surtout dans des conditions de vie réelle, à domicile.
Pour autant, l’app montre quelques limitations : l’étude n’a porté que sur quelques semaines et l’outil ne s’adresse qu’à des patients SMA âgés de plus de six ans et donc capables de manipuler un téléphone portable. Elle ne peut remplacer les évaluations par des professionnels de santé en milieu spécialisé et vient simplement en complément. On peut aussi imaginer qu’au long cours, des phénomènes de lassitude liée à l’utilisation d’une telle application numérique risquent de se produire.
Un modèle in vitro destiné à tester des thérapies innovantes dans l’amyotrophie spinale et les pathologies liées au gène IGHMPB2
In vitro modeling as a tool for testing therapeutics for spinal muscular atrophy and IGHMBP2-related disorders
Actualité commentée réalisée par Jon Andoni Urtizberea - Publiée le 11 septembre 2023
TKMR-N 2023 ; 2 (18) : A18
Sierra-Delgado JA, Sinha-Ray S, Kaleem A, Ganjibakhsh M, Parvate M, Powers S, et al.
Biology 2023 ; 12 : 867.
AVIS D’EXPERT
Les modèles cellulaires ou animaux sont toujours utiles à la compréhension d’une maladie neuromusculaire d’origine génétique. Si le muscle est relativement facile à modéliser à partir de cellules souches musculaires, telles les cellules satellites, il n’en est pas de même pour les motoneurones dont le corps cellulaire est situé dans la corne antérieure de la moelle épinière. Les chercheurs démontrent dans l’article cité ici en référence qu’on peut utiliser une méthode simple et rapide pour remédier à ces difficultés. La conversion directe consiste à reprogrammer des fibroblastes à l’aide d’un cocktail de petites molécules sans passer par l’étape, longue et fastidieuse, de l’obtention préalable de cellules souches reprogrammées (Induced Pluripotent Stem Cells ou IPSc). Les limites de l’utilisation des IPSc sont désormais bien connues : problèmes de maintenance, grande variabilité dans les clones cellulaires obtenus ou perte de marqueurs épigénétiques.
Le deuxième enseignement de ces travaux réside dans l’observation de phénotypes clairement pathologiques lorsque les neurones transformés proviennent de patients atteints de pathologies dégénératives du motoneurone. Les anomalies étaient observables en microscopie à l’aide de marqueurs fluorescents et en Western blot.
De manière originale, les chercheurs ont enfin pu démontrer, avec ce nouveau modèle, le bénéfice des thérapies de transfert de gène dans les deux types de pathologies.
Dans le sens inverse, et plus dans un but diagnostic, cette nouvelle méthode a permis, pour la première fois, d’affirmer avec certitude le caractère pathologique d’un variant jusque-là considéré comme sans véritable signification (Variant of Unknown Significance ou VUS). La conversion directe et l’observation d’un phénotype motoneuronal dégénératif pourraient ainsi lever bien des ambiguïtés, surtout dans les pathologies liées au gène IGHMPB2. La réponse à la thérapie génique telle qu’observée dans ce cas précis serait un argument supplémentaire de pathogénicité.
La principale limitation de la conversion directe tient au fait que les fibroblastes ainsi transformés produisent habituellement plus des neurones de type gabaergiques et glutamatergiques que des motoneurones ou des neurones cholinergiques. Malgré cela, les phénotypes observés ici sont clairement de type motoneuronaux.
Le rôle crucial de la titine dans le développement fœtal : fausses couches et anomalies variées des muscles, des os et du cœur caractérisent les formes les plus graves de titinopathie
The crucial role of titin in fetal development: recurrent miscarriages and bone, heart and muscle anomalies characterise the severe end of titinopathies spectrum
Actualité commentée réalisée par Jon Andoni Urtizberea - Publiée le 04 septembre 2023
TKMR-N 2023 ; 2 (17) : A17
Di Feo MF, Lillback V, Jokela M, McEntagart M, Homfray T, Giorgio E, et al.
J Med Genet 2023 ; 0 : 1-8.
AVIS D’EXPERT
D’abord décrites chez l’adulte dans le contexte des myopathies des ceintures ou des myopathies fibrillaires, les titinopathies ont vu leur spectre phénotypique considérablement s’élargir ces dernières années, notamment en direction de la population pédiatrique. Dans l’étude citée en référence et consacrée aux formes les plus précoces et les plus sévères de la maladie, on mesure mieux l’ampleur et la prévalence du phénomène. Les titinopathies représentent en effet une cause beaucoup plus fréquente de morbi-mortalité périnatale d’origine musculaire et/ou cardiaque que ce que l’on pensait. Ceci provient du fait que le gène TTN est de très grande taille et que les variations de séquence en son sein restaient jusqu’ici d’interprétation délicate. Ceci est désormais moins vrai avec les variants tronquants (TTNtv) dont le caractère pathogène est pratiquement toujours vérifié. De plus, l’établissement de corrélations plus claires entre le génotype et le phénotype permet au clinicien de mieux apprécier la gravité de la situation.
Ces travaux pourraient avoir une incidence directe sur la façon dont sont explorés les syndromes d’akinésie fœtale ou de syndromes polymalformatifs, notamment dans le cadre du conseil génétique. Le caryotype et la recherche d’une pathologie micro-délétionnelle par CGH-array restent les examens standards dans de tels contextes mais ne sont pas en mesure de détecter les anomalies, plus subtiles, du gène TTN, lesquels requièrent du séquençage.
Pour autant, l’interprétation de ces variants TTNtv reste parfois délicate notamment chez les apparentés qui en sont porteurs à l’état hétérozygote. Certains d’entre eux développent une cardiomyopathie sans qu’un lien de causalité n’ait été clairement établi.
Enfin, et comme le reconnaissent les auteurs, les données issues de la seule littérature (pour les 93 cas recensés) sont souvent incomplètes et de qualité inégale. Ce qui n’enlève rien, à notre sens, au message délivré dans cet article remarquable.
Cas clinique : caractérisation clinique et moléculaire d’une fratrie atteinte de myopathie de Brody
Case report: Clinical and molecular characterization of two siblings affected by Brody myopathy
Actualité commentée réalisée par Jon Andoni Urtizberea - Publiée le 28 août 2023
TKMR-N 2023 ; 2 (16) : A16.
Velardo D, Antognozzi S, Rimoldi M, Pagliarani S, Cogiamanian F, Barbieri S, et al.
Front Neurol 2023 ; 12 : 117071.
AVIS D’EXPERT
Pour très rare qu’elle soit, la myopathie de Brody n’en est pas moins intéressante. Sa symptomatologie s’apparente à celle observée dans les myotonies, que celles-ci soient dystrophiques ou non. Les patients atteints de myopathie de Brody se plaignent en effet de raideur musculaire, ce qui est la traduction d’un défaut de relaxation musculaire. Cette raideur est souvent en lien avec l’exercice physique mais sans que l’on observe de phénomène de warm-up. Très souvent, le diagnostic de myotonie est évoqué à l’interrogatoire mais est ensuite infirmé du fait de la négativité de l’examen clinique (il n’y a pas de myotonie détectable au niveau des mains ou des autres muscles) et des examens complémentaires. Il n’y a pas, dans la myopathie de Brody, de décharges myotoniques spontanées ou induites par l’effort. A défaut de penser à une myopathie de Brody, d’autres diagnostics différentiels sont fréquemment évoqués, comme les différentes formes de neuromyotonie ou une pathologie fonctionnelle. La notion d’autres sujets atteints dans la famille représente toutefois un indice d’organicité et doit inciter à poursuivre les investigations jusqu’au test génétique de confirmation.
L’observation rapportée dans l’article cité en référence n’est pas exceptionnelle en soi. Le phénotype est comparable à celui rapporté dans une série de 40 patients atteints de myopathie de Brody tout comme la durée de l’errance diagnostique (ici, plus d’une dizaine d’années)1. La notion de contractures silencieuses à l’EMG n’est pas toujours présente mais peut constituer un bon élément d’orientation comme ici. In fine, et comme dans de plus en plus de pathologies neuromusculaires difficiles à caractériser et/ou connues, le séquençage à haut débit fait la preuve, une fois de plus, de sa très grande efficacité. Ici, les patients étaient porteurs tous deux d’une délétion du gène ATP2A1 déjà connue (c.324 + 1G > A) et d’un nouveau variant de séquence très vraisemblablement pathologique au regard du contexte (c.324 + 1G > A).
Même s’il n’existe pas de traitement spécifique pour cette forme de myopathie, la reconnaitre tôt évite l’errance et permet de rassurer les patients sur le caractère bénin de l’affection et sur leur devenir fonctionnel.
Un cas de syndrome d’Andersen-Tawil confondu avec une dystrophie musculaire
A case report of Andersen-Tawil syndrome misdiagnosed with myodystrophy
Actualité commentée réalisée par Jon Andoni Urtizberea - Publiée le 22 août 2023
TKMR-N 2023 ; 2 (15) : A15
Zhao X, Zu H, Yao K.
Front Neurol 2023 ; 14 : 1170693.
AVIS D’EXPERT
Le syndrome d’Andersen-Tawil est une pathologie neuromusculaire ultra-rare classée parmi les paralysies périodiques. Des mutations dans le gène KCNJ2 en sont à l’origine, celui-ci codant un canal potassium également référencé sous le nom de kir2.1. Les anomalies de fonctionnement de ce canal entrainent une hyperexcitabilité membranaire, tant au niveau du muscle squelettique que du cœur. Le diagnostic est difficile à poser du fait de modes d’entrée dans la maladie très variés (épisodes paralytiques, troubles du rythme à type d’arythmie ventriculaire ou de QT-long, autres) et d’un syndrome malformatif (dysmorphie faciale ou déformations squelettiques) souvent difficile à reconnaitre.
Dans le cas présent, le patient présentait une histoire relativement typique d’ATS au début de la maladie. A cette époque, toutefois, le syndrome n’avait pas encore été décrit ; il ne le sera qu’en 19941. La survenue progressive d’un déficit musculaire permanent a également été rapportée dans ce type de paralysie périodique, au point de mimer une pathologie musculaire dégénérative avec augmentation sensible des CPK.
Les auteurs rappellent, à juste titre, tous les pièges qui peuvent conduire à des errances diagnostiques dans le contexte de l’ATS. C’est dire l’importance, également rappelée ici, d’un interrogatoire bien conduit à la recherche d’épisodes paralytiques et d’un examen physique minutieux à la recherche de malformations squelettiques et faciales souvent subtiles.
Enfin, le fait de faire un diagnostic d’ATS précocement permet de prévenir les accidents d’anesthésie, lesquels sont relativement fréquents dans cette pathologie et de proposer une prise en charge adaptée pour les troubles du rythme cardiaque.
Sécurité, tolérance et pharmacocinétique de l’éteplirsen chez des jeunes garçons (6 à 48 mois) atteints de dystrophie musculaire de Duchenne et éligibles au saut de l’exon 51
Safety, tolerability and pharmacokinetics of eteplirsen in young boys aged 6–48 months with Duchenne muscular dystrophy amenable to exon 51 skipping
Actualité commentée réalisée par Jon Andoni Urtizberea - Publiée le 19 juillet 2023
TKMR-N 2023 ; 2 (14) : A14
Mercuri E, Seferian AM, Servais L, Deconinck N, Stevenson H, Ni X, et al.
Neuromuscul Disord 2023 ; 33 : 476-83.
AVIS D’EXPERT
Le saut d’exon thérapeutique (ou SET) est une méthode de thérapie génique reposant sur des oligonucléotides anti-sens (OAN pour antisense oligonucleotides). Les AON sont des molécules chimiques qui jouent sur l’épissage du gène DMD et qui permettent de restaurer le cadre de lecture en shuntant un exon donné. In fine, cette approche thérapeutique aboutit à la production d’une dystrophine en grande partie fonctionnelle. La myopathie de Duchenne se prête bien au SET du fait de la présence de nombreuses délétions au sein du gène DMD. L’exon 51 reste la cible de choix car permettant de traiter entre 12 et 15 % de la population des patients atteints de DMD.
L’éteplirsen fait office de pionnier en la matière. Cet AON développé par le laboratoire américain Sarepta Therapeutics et visant précisément l’exon 51, a été approuvé par la FDA sur la base d’essais cliniques concluants dans une population d’enfants DMD âgés de plus de quatre ans. Pour autant, l’éteplirsen n’est ni approuvé ni commercialisé en Europe. Les données de vie réelle en provenance des USA semblent confirmer l’efficacité déjà observée avec un profil de sécurité toujours aussi acceptable, à la différence du drisapersen, le tout premier AON développé par le laboratoire Biomarin mais dont la toxicité rénale et cutanée avait empêché l’obtention de l’autorisation de mise sur le marché par la FDA.
L’étude citée en référence est intéressante à plus d’un titre. Elle confirme la bonne tolérance du produit, mais cette fois-ci dans une population d’enfants DMD beaucoup plus jeunes. Ceci pourrait ainsi ouvrir la voie à des traitements précoces dans le cadre, ou non, d’un dépistage néonatal de la DMD. Le sujet du dépistage reste toutefois largement débattu, aux USA comme en Europe. Le fait que l’essai clinique cité en référence ait eu lieu en Europe apporte également la preuve que le laboratoire Sarepta Therapeutics continue à s’intéresser au marché du Vieux Continent. L’essai a ainsi permis à un certain nombre de patients européens atteints de DMD de bénéficier de ce produit, au moins pour la durée de l’étude.
Enfin, la question de la place de l’éteplirsen par rapport à la nouvelle génération d’AON (PPMO, tricyclo-AON, etc.) en cours de développement clinique reste posée, tout comme son avantage sélectif vis-à-vis des thérapies géniques à base de micro-dystrophine dont le premier produit, aussi développé par Sarepta Therapeutics, vient d’être approuvé aux USA de manière conditionnelle.
Signatures moléculaires des formes héréditaires et acquises (de début tardif) de myopathie à bâtonnets
Molecular signatures of inherited and acquired sporadic late onset nemaline myopathies
Actualité commentée réalisée par Jon Andoni Urtizberea - Publiée le 05 juillet 2023
TKMR-N 2023 ; 2 (13) : A13
Nicolau S, Dasgupta A, Dasari S, Charlesworth MC, Johnson KL, Pandey AL, et al.
Acta Neuropathol Commun 2023 ; 11 : 20.
AVIS D’EXPERT
Les bâtonnets ou nemalin bodies (du grec nêma pour bâton) sont des petites structures pathologiques observables en microscopie optique mais encore mieux caractérisées en microscopie électronique. Ils ont été d’abord décrits dans des formes très précoces, et souvent très sévères, de myopathie congénitale génétiquement déterminée. On compte à ce jour une quinzaine de gènes responsables de myopathies à bâtonnets à caractère héréditaire et la liste n’est sans doute pas close. On trouve toutefois des bâtonnets dans d’autres pathologies musculaires, héréditaires ou non, ce qui a toujours fait relativiser leur caractère spécifique. La découverte de la SLONM en 1966 est venue illustrer cette grande diversité, tout comme dans les infections à VIH quelques années plus tard. La SLONM est classiquement décrite comme associant une myopathie de gravité variable, mais toujours de début tardif, et une gammapathie monoclonale. Il arrive que la gammapathie manque au tableau, d’où la nécessité de mieux affiner les critères diagnostiques de la SLOMN. C’est en grande partie l’objectif que se sont fixés les investigateurs dans le travail cité en référence. Ceci est d’autant plus important que la SLONM, à la différence des formes héréditaires, peut bénéficier d’une immunothérapie dont le résultat est parfois spectaculaire. L’originalité des travaux présentés ici tient dans la combinaison des approches utilisées, qu’il s’agisse de l’histologie musculaire et surtout de la protéomique et de la transcriptomique appliquées au tissu musculaire. Ceci est désormais possible du fait d’une meilleure gestion à grande échelle de multiples données biologiques (big data). On connait désormais mieux les mécanismes, très distincts en formes acquises et héréditaires , de formation desdits bâtonnets. La principale limitation de l’étude tient au fait qu’il s’agit de méthodes difficilement transposables en routine du fait de leur lourdeur. On reste clairement dans le domaine de la recherche. Ceci est d’autant plus vrai qu’on pratique de moins en moins de biopsies musculaires à visée diagnostique et que les experts du domaine se font de plus en plus rares.
L’onasemnogene abeparvovec dans l’amyotrophie spinale : des facteurs prédictifs d’efficacité et d’innocuité chez des patients naïfs et chez des patients ayant bénéficié d’un switch thérapeutique
Onasemnogene abeparvovec in spinal muscular atrophy: predictors of efficacy and safety in naïve patients with spinal muscular atrophy and following switch from other therapies
Actualité commentée réalisée par Jon Andoni Urtizberea - Publiée le 14 juin 2023
TKMR-N 2023 ; 2 (12) : A12
Pane M, Berti B, Capasso A, Coratti G, Varone A, D'Amico A, et al.
EClinicalMedicine 2023 ; 59 : 101997.
AVIS D’EXPERT
Les données de vie réelle sont souvent plus pertinentes que les données issues des essais cliniques. La SMA ne fait pas exception. Dans cette étude portant sur des patients traités en Italie par le seul produit de transfert de gène autorisé dans la SMA, on voit bien que les indications ont largement évolué par rapport aux autorisations initiales de mise sur le marché. Les prescripteurs locaux traitent des enfants soit franchement plus tôt, dans le cadre d’un dépistage néonatal ou non, ou plus tard, bien au-delà de la limite classique des 24 mois. De même concernant le critère de poids dont certains prescripteurs s’affranchissent également. On note également qu’un nombre non négligeable d’enfants reçoivent du nusinersen avant de switcher vers l’OA. Les motifs de switch pouvaient être variés : soit il s’agissait d’un traitement transitoire d’attente soit le patient était en échec thérapeutique avec le nusinersen. La cohorte étudiée ici est imposante par sa taille (67 enfants inclus), ce qui témoigne d’une diffusion très large de l’OA comme traitement de première intention, surtout chez les tout-petits. L’autre enseignement réside dans la constatation de nombreux effets secondaires comme l’ont déjà rapporté d’autres équipes utilisant la technologie de transfert de gène, à commencer par l’élévation des enzymes hépatiques. Fort heureusement, aucun des deux décès observés dans la cohorte n’a été imputé au traitement. L’intérêt du dépistage néonatal et donc d’un traitement précoce, est de nouveau souligné. Ceci est très particulièrement net sur les données d’efficacité. En revanche, les modèles statistiques utilisés ne permettent pas d’évaluer les éventuels bénéfices liés à la combinaison des trois drogues. On notera enfin que, même traités au-delà des 24 mois, les enfants peuvent être améliorés sur le plan fonctionnel à condition de juger cela non pas sur le court terme (6 mois), mais plutôt sur le long terme (au-delà de 12 mois).
L’apport du gène de la micro-dystrophine améliore significativement la fonction musculaire et l’histologie du diaphragme dans un modèle murin fibrotique de la dystrophie musculaire de Duchenne
Microdystrophin gene addition significantly improves muscle functionality and diaphragm muscle histopathology in a fibrotic mouse model of Duchenne muscular dystrophy
Actualité commentée réalisée par Jon Andoni Urtizberea - Publiée le 02 juin 2023
TKMR-N 2023 ; 2 (11) : A11
Cernisova V, Lu-Nguyen N, Trundle J, Herath S, Malerba A, Popplewell L.
Int J Mol Sci 2023 ; 24 : 8174.
AVIS D’EXPERT
Les chercheurs ont utilisé le modèle murin D2.mdx dont le phénotype, à la différence de la souris mdx originale, se rapproche fortement de la pathologie humaine, particulièrement au niveau de la fibrose musculaire. De tels modèles, tout comme celui développé il y a peu chez le rat, sont très utiles pour le développement des thérapies. Les travaux cités en référence ne constituent pas une vraie première mais confirment l’intérêt et l’efficacité du transfert thérapeutique de gène dans la myopathie de Duchenne. Ils sont même très encourageants quant aux bénéfices obtenus au niveau clinique. On notera que des données, en partie similaires, avaient été précédemment rapportées en utilisant le vecteur AAV9 dans la souris modèle D2.mdx, à la fois pour des transgènes de type micro-dystrophine ou de type micro-utrophine. Le construct micro-dystrophine choisi ici est le plus structurellement proche parmi ceux utilisés en clinique, et en particulier celui de la micro-dystrophine mis au point par le laboratoire Généthon et testé depuis peu chez plusieurs malades. L’autre particularité de ce transgène est qu’il nécessite des doses moindres de virus (4 x 1012 vg). De ce fait, et à efficacité comparable, il est moins immunogène. Les auteurs ne signalent d’ailleurs pas de problème de toxicité liée à la méthode utilisée. L’efficacité sur la fibrose musculaire, un des processus majeurs à l’origine de la dégénérescence observée dans la DMD est ici remarquable, même si les auteurs se sont cantonnés à l’étude du diaphragme. On aurait bien aimé avoir des données d’efficacité aussi sur le muscle cardiaque, un autre organe cible dans la DMD. On peut aussi noter que le nombre total de souris étudiées n’est pas très élevé, ce qui n’a pas empêché d’obtenir des preuves statistiquement valides. Ces travaux confirment l’intérêt de la thérapie par addition de gène dans la dystrophie musculaire de Duchenne. On peut légitimement espérer qu’ils seront, en terme d’efficacité, transposables chez l’homme comme semblent le démontrer les essais cliniques en cours.
Des repères microscopiques et biochimiques dans l’histologie musculaire associée à des nouveaux variants du gène BICD2
Microscopic and biochemical hallmarks of BICD2-associated muscle pathology toward the evaluation of novel variants
Actualité commentée réalisée par Jon Andoni Urtizberea - Publiée le 15 mai 2023
TKMR-N 2023 ; 2 (10) : A10
Unger A, Roos A, Gangfuß A, Hentschel A , Gläser D, Krause K, et al.
Int J Mol Sci 2023 ; 24 : 6808.
AVIS D’EXPERT
Les études NGS (panel de gènes ou exome entier selon le cas) qui sont réalisées chez des patients neuromusculaires en errance diagnostique mettent parfois en évidence des variants de séquence du gène BICD2, sans que leur pathogénicité puisse être formellement établie. Ce gène code une protéine impliquée, notamment dans le motoneurone, dans le trafic intracellulaire entre les vésicules membranaires du Golgi et du réticulum endoplasmique, et la membrane plasmique. Au niveau du premier motoneurone, les mutations de BICD2 entraînent une paraplégie spastique héréditaire. Au niveau de la moelle épinière, le phénotype est celui d’une amyotrophie spinale avec une prédominance des signes au niveau des membres inférieurs (SMA-LED2 pour amyotrophie spinale avec prédominance au niveau des membres inférieurs de type 2). Comme dans d’autres pathologies du motoneurone, il existe un continuum entre ces deux phénotypes. On notera aussi que DYNC1H1, le premier gène responsable du phénotype SMA-LED1 code une protéine impliquée dans le complexe dynéine-dynactine avec lequel le produit du gène BICD2 interagit étroitement par l’intermédiaire de la protéine RAB6A. L’existence d’un phénotype musculaire associé à BICD2 se révèle donc intéressante d’un point de vue physiopathologique et semble ne pas relever de l’exception. Ceci est corroboré par les signes, à la fois neuropathique et myopathique, observés chez la souris transgénique invalidée pour le gène BICD2. D’après les auteurs, la localisation des mutations à l’intérieur du gène BICD2 expliquerait en partie la variabilité de l’expression phénotypique. Les auteurs font l’hypothèse que l’atteinte motoneuronale pourrait être secondaire à une perturbation primitive du muscle, la surexpression de la thrombospondine-4 et du biglycane allant dans ce sens. Ce même genre de débat a existé aussi pour expliquer la genèse de l’amyotrophie spinale classique liée au gène SMN1. Pour autant, cette étude présente quelques insuffisances. L’existence, de manière concomitante, de variants dans d’autres gènes connus pour donner des myopathies (COL6A1 et FLNC) vient compliquer l’interprétation des données. On aurait également aimé disposer des données électrophysiologiques de chaque patient.
L’évaluation de cellules souches humaines pluripotentes issues d’un patient atteint du syndrome de Schwartz-Jampel met au jour une hyperexcitabilité particulière des muscles squelettiques
Evaluation of human-induced pluripotent stem cells derived from a patient with Schwartz–Jampel syndrome revealed distinct hyperexcitability in the skeletal muscles
Actualité commentée réalisée par Jon Andoni Urtizberea - Publiée le 03 mai 2023
TKMR-N 2023 ; 2 (9) : A9
Yamashita Y, Nakada S, Nakamura K, Sakurai S, Ohno K, Goto T, et al.
Biomedicines 2023 ; 11 : 814.
AVIS D’EXPERT
Le perlécan, un constituant de la matrice extra-cellulaire mis en cause dans le SJS, est une protéine de très grande taille avec, de ce fait, de multiples interactions avec d’autres molécules. Le perlécan intervient ainsi dans de nombreux processus cellulaires, que ce soit au niveau du muscle ou du cartilage. A la différence des dystrophies myotoniques non dystrophiques comme la maladie de Thomson (autosomique dominante) ou la maladie de Becker (autosomique récessive), le SJS n’est pas lié directement au dysfonctionnement d’un canal ionique musculaire mais à celui d’une protéine de structure. Certains auteurs attribuent l’hyperexcitabilité musculaire à la terminaison nerveuse ou à la jonction neuromusculaire, et n’hésitent pas à classer le SJS parmi les syndromes neuromyotoniques au même titre que, par exemple, le syndrome d’Isaacs ou la neuromyotonie liée au gène HINT1. Les chercheurs japonais à l’origine des travaux présentés ici se sont concentrés sur le volet neuromusculaire du syndrome de Schwartz-Jampel en créant des cellules souches pluripotentes (hiPSC) d’un individu atteint d’une forme classique de SJS, avec deux variants hétérozygotes composites du gène HSPG2. Ils ont pu démontrer, grâce à une technique de mesure des mouvements calciques intracellulaires – technique utilisée dans d’autres modèles et donc reproductible – que le modèle créé, en l’occurrence des myotubes dérivés de ces hiPSC, présentaient bien des troubles de l’excitabilité membranaire avec une sensibilité accrue à l’acétylcholine. Ces cellules n’étant pas encore, par définition, innervées, les auteurs font l’hypothèse que la composante nerveuse de l’hyperexcitabilité cellulaire observée dans le SJS est probablement minime voire inexistante. Le modèle cellulaire ainsi mis au point pourra également servir au développement de thérapeutiques dans le SJS en permettant de cribler toute une série de molécules, en plus de celles déjà utilisées en clinique. Dans une certaine mesure, il pourrait remplacer l’examen électrophysiologique en clinique, lequel se révèle non seulement invasif mais compliqué à interpréter chez les très jeunes enfants. Le fait que les lignées cellulaires soient issues ici d’un seul patient limite un peu la portée de cette étude mais ne remet pas en cause la pertinence des résultats. De manière plus générale, ces travaux soulignent l’intérêt des cellules souches pluripotentes comme moyen de modéliser, à moindre frais et dans de bonnes conditions de reproductibilité, certaines maladies génétiques mendéliennes.
Une coupure unique par le système Cas9 permet la restauration, sans nécessité de template, d’une mutation fondatrice responsable d’une forme de dystrophie musculaire
Cas9-induced single cut enables highly efficient and template-free repair of a muscular dystrophy causing founder mutation
Actualité commentée réalisée par Jon Andoni Urtizberea - Publiée le 17 avril 2023
TKMR-N 2023 ; 2 (8) : A8
Müthel S, Marg A, Ignak B, Kieshauer J, Escobar H, Stadelmann C, et al.
Mol Ther Nucleic Acids 2023 ; 31 : 494-511.
AVIS D’EXPERT
Les calpaïnopathie primaires sont des myopathies des ceintures à transmission majoritairement autosomique récessive et sont le résultat de mutations dans le gène CAPN3. Le déficit musculaire induit par ces mutations débute au cours de la deuxième décennie et conduit à une perte de la marche au bout de 20 à 30 ans d’évolution de la maladie. CAPN3 code une enzyme jouant un rôle dans la maintenance et le remodelage sarcomériques. La protéine a une forte activité autolytique ce qui explique sa dégradation rapide lors des études in vitro et les difficultés à disposer de modèles animaux pertinents. Les thérapies innovantes ciblant cette pathologie n’en sont qu’au stade préclinique. La thérapie de remplacement génique par virus associé à l’adénovirus (AAV) s’est avérée bénéfique et efficace chez la souris et chez les primates non-humains. Les travaux présentés ici constituent une preuve de concept supplémentaire de l’intérêt de la thérapie génique combinant édition génomique et thérapie cellulaire1. Cette approche novatrice dont le fer de lance est constitué par le désormais célèbre système CRISP-Cas9 permet, très schématiquement, de corriger tout type d’anomalie d’un gène donné, et jusqu’à l’échelle d’une seule base nucléotidique. Les expérimentations d’édition génomique commencent à se mettre en place chez l’homme dans d’autres pathologies héréditaires et semblent prometteuses malgré un risque d’effets secondaires qui reste à déterminer. A juste titre, les auteurs ont choisi de tenter de réparer le défaut moléculaire le plus fréquemment rencontré car étant le résultat d’un très probable effet fondateur. Il s’agissait en l’occurrence d’une délétion d’une seule base (c.550delA) entrainant un décalage du cadre de lecture. L’originalité de ces travaux tient au choix du type d’édition génomique (car ne nécessitant pas de template), au fait qu’il s’agissait de modifier une seule base et aux perspectives qu’ils ouvrent en termes de thérapie cellulaire. On pourrait effectivement envisager une application clinique de cette approche de correction ex vivo par des transplantations autologues d’IPSC modifiées. C’est d’ailleurs ce à quoi les auteurs de l’article s’attèlent désormais (protocole Eudra-CT no. 2021-002004-13). Les limitations d’une telle méthode sont toutefois réelles. Le nombre modeste de cellules complètement éditées pourraient, par exemple, ne pas se traduire par une amélioration clinique. L’expérience prouve que les greffes de cellules dans le muscle sont presque toujours décevantes. Seul un essai thérapeutique, même conduit en ouvert, pourra en apporter la preuve.
Une atteinte des muscles des ceintures est causée par des mutations du gène HMGCR et la myopathie liée aux statines est traitable par la mévalonolactone
Limb girdle muscular disease caused by HMGCR mutation and statin myopathy treatable with mevalonolactone
Actualité commentée réalisée par Jon Andoni Urtizberea - Publiée le 05 avril 2023
TKMR-N 2023 ; 2 (7) : A7
Yogev Y, Shoerer Z, Koifman A, Wormser O, Drabkin M, Halperin D et al.
PNAS 2023 ; 7 : e2217831120.
AVIS D’EXPERT
Les travaux cités en référence constituent une première. La forme auto-immune de déficience en HMG-CoA réductase a été décrite par l’équipe d’Andrew Mammen en 20111 et son diagnostic repose sur la positivité d’auto-anticorps dirigés contre cette enzyme. Initialement considérée comme causée par une exposition aux statines (des médicaments hypocholestérolémiants très largement prescrits), cette myopathie a vu progressivement son champ phénotypique s’étendre, notamment chez les enfants. Le fait d’identifier une mutation récessive, confirmée comme pathogène par les études fonctionnelles, dans le gène HMGCR codant l’HMG-CoA réductase représente une avancée importante dans notre compréhension de la physiopathologie des maladies musculaires en rapport avec cette voie métabolique. Tout aussi importants sont les efforts des chercheurs pour imaginer, à partir de leur découverte, une approche thérapeutique ciblée. Le traitement par mévalonolactone parait simple, facile d’administration, et surtout, efficace à en juger par l’évolution des signes cliniques, tant chez le patient considéré que chez l’animal modèle de la myopathie iatrogène liée aux statines. L’étude n’est toutefois pas exempte de limitations. Il s’agit, à ce jour, d’une observation unique, au sein d’une seule famille caractérisée par une grande endogamie. Cette découverte prendra plus de valeur lorsque d’autres équipes à travers le monde auront, on peut le souhaiter, rapporté des cas familiaux similaires. Enfin, les conditions de l’essai thérapeutique sont très discutables, ce dernier n’ayant été conduit que chez un seul malade, avec une observance rapportée comme limitée, et un composé chimique dont la toxicité n’avait pas été préalablement testée. Les conclusions découlant des observations cliniques, pour intéressantes qu’elles soient, sont donc à analyser avec précaution, aussi bien chez l’homme que chez l’animal. La piste de la mévalonolactone reste néanmoins intéressante et pourrait s’avérer très utile pour les cas de MNAI. Dans cette dernière, l’arsenal thérapeutique repose sur un traitement lourd et prolongé à base d’immunosuppresseurs et, le cas échéant, de corticoïdes. Le mévalonolactone pourrait donc, si sa faible toxicité et son efficacité étaient confirmées, constituer une alternative ou, plus vraisemblablement, un complément au traitement conventionnel.
Le double coup dur de DUX4 : le facteur de transcription causant une dystrophie musculaire rare tue aussi les cellules progénitrices du nez chez l’homme
DUX4 double whammy: The transcription factor that causes a rare muscular dystrophy also kills the precursors of the human nose
Actualité commentée réalisée par Jon Andoni Urtizberea - Publiée le 15 mars 2023
TKMR-N 2023 ; 2 (6) : A6
Inoue K, Bostan H, Browne MR, Bevis OF, Bortner CD, Moore SA, et al.
Sci Adv 2023 ; 9 : eabq7744.
AVIS D’EXPERT
La FSHD est une des myopathies les plus fréquentes chez l’adulte. Elle entraine un déficit musculaire très sélectif, souvent progressif, avec une grande variabilité phénotypique inter et intra-familiale. Elle existe sous deux formes selon que l’on observe une perte de matériel génétique dans la région D4Z4 ou une mutation ponctuelle dans le gène SMCHD1. Dans les deux cas, on note une réexpression de DUX4, un facteur de transcription censément mis au repos après les premières phases du développement embryonnaire. L’hypométhylation de la région jouerait un grand rôle dans la survenue de la FSHD. L’arhinie est quant à elle une malformation congénitale très rare de l’appendice nasal. Il a été démontré que des mutations du gène SMCHD1, dont une particulièrement fréquente, étaient impliquées dans le processus malformatif. A mutation de SMCHD1 identique, il existe toutefois une assez grande variabilité phénotypique (formes mineures, formes incomplètes) suggérant l’existence, tout comme dans la FSHD, de facteurs épigénétiques. Les chercheurs ont réussi ici à faire le lien entre les deux syndromes bien que cliniquement, ils n’aient rien en commun. Dans les deux cas, la toxicité cellulaire de DUX4 a été confirmée. Cela vaut pour les fibres musculaires comme pour certaines des cellules progénitrices du nez. Parmi elles, seules les cellules de placodes sont exposées et sensibles à la toxicité de DUX4, à la différence des cellules de la crête neurale. Pour expliquer la variabilité phénotypique, les chercheurs mettent en avant l’existence d’autres facteurs : soit des gènes modificateurs liés à l’individu lui-même, soit des facteurs environnementaux. Cette étude permet de progresser dans la compréhension des processus très complexes de différenciation cellulaire chez l’embryon, notamment dans la formation des organes sensoriels. De ce point de vue, l’arhinie serait le premier exemple d’une malformation cranio-faciale expliquée par une dérépression d’origine épigénétique d’une protéine toxique. Elle permet aussi de confirmer le rôle central du facteur de transcription DUX4 dans la physiopathologie de la FSHD. Ce rôle a été critiqué, à tort, pendant plusieurs années.
Un cas de myopathie myofibrillaire hypertonique avec issue fatale chez un nourrisson avec mutations hétérozygotes composites du gène CRYAB
Case report: Fatal infantile hypertonic myofibrillar myopathy with compound heterozygous mutations in the CRYAB gene
Actualité commentée réalisée par Jon Andoni Urtizberea - Publiée le 02 mars 2023
TKMR-N 2023 ; 2 (5) : A5
Zhang SS, Gu LN, Zhang T, Xu L, Wei X, Chen SH, et al.
Front Ped 2023 ; 10 : 993165.
AVIS D’EXPERT
Les myopathies myofibrillaires sont des pathologies neuromusculaires rares touchant préférentiellement les adultes et se traduisant par un déficit moteur progressif, volontiers distal, et des troubles cardiaques. Elles sont causées par moins d’une dizaine de gènes, le plus souvent autosomiques dominants (DES, CRYAB, MYOT, FLNC, ZASP, BAG3). L’observation présentée ici est intéressante à plus d’un titre. Elle démontre d’abord que certains cas de myopathie myofibrillaire peuvent se manifester dans les premiers mois de vie avec un pronostic vital rapidement engagé du fait d’une atteinte respiratoire sévère. Ces formes précoces, voire ultra-précoces, sont, sous réserve d’inventaire, l’apanage des mutations du gène CRYAB. Celui-ci code l’alpha-B-cristalline et donne classiquement une myopathie de l’adulte associée à une cataracte. Les premiers cas de cette nouvelle entité résumée par l’acronyme FIHMM (pour Fatal Infantile Hereditary Myofibrillar Myopathy) ont été rapportés chez les aborigènes canadiens. Dans les observations qui ont suivi, le tableau clinique s’enrichit d’une hypertonie hypertrophique des muscles abdominaux très inhabituelle et dont la signification est discutée. Les études électrophysiologiques ne sont d’ailleurs pas en faveur d’un processus myotonique mais bien d’une atteinte primitivement myogène. Les taux de CPK sont normaux ou légèrement augmentés. L’autre intérêt de ce cas réside dans le génotype observé dans cette famille. Au vu du tableau clinique et de l’histologie musculaire, les critères sont réunis pour affirmer avec certitude le diagnostic de myopathie myofibrillaire liée à CRYAB. Le premier variant est très vraisemblablement causal bien qu’inconnu dans les bases de données NGS. Le variant le plus pathogène (p.Met1Ile), quant à lui, a été rapporté à maintes reprises dans des cas pédiatriques en Chine, laissant d’ailleurs supposer l’existence d’un effet fondateur dans la population Han majoritaire dans le pays. Reste que le pronostic de cette forme du nourrisson reste globalement très mauvais, l’enfant présentée ici étant décédée peu de temps après son admission et suite à l’arrêt des soins de réanimation.
Une approche alliant recherche du gène et expression de gènes-candidats permet d’améliorer le diagnostic du syndrome d’Escobar
Combining gene mutation with expression of candidate genes to improve diagnosis of Escobar syndrome
Actualité commentée réalisée par Jon Andoni Urtizberea - Publiée le 13 février 2023
TKMR-N 2023 ; 2 (4) : A4
Najjar D, Chikhaoui A, Zarrouk Z, Azouz S, Kamoun W, Nassib N, et al.
Genes 2022 ; 13 : 1748-66.
AVIS D’EXPERT
Le syndrome d’Escobar est une pathologie génétique ultra-rare qu’il faut savoir évoquer dans un contexte clinique d’arthrogrypose multiple à révélation précoce. Il est vraisemblablement le résultat d’une akinésie fœtale d’origine neuromusculaire. Il existe quelques variantes sur le plan clinique, certains traits, notamment dysmorphiques, étant moins marqués et donc plus difficiles à diagnostiquer à la naissance. Ceci explique les quelques cas où le diagnostic a pu être porté avec retard. La prise en charge est complexe et repose sur le traitement orthopédique et/ou chirurgical des déformations articulaires du tronc (scoliose) et des membres (flessums). Ces déformations peuvent également interférer avec la fonction respiratoire. De manière générale, on associe volontiers ce syndrome aux maladies neuromusculaires à expression très précoce du fait de l’implication d’au moins deux gènes à expression neuromusculaire : celui codant la sous-unité gamma du récepteur à l’acétylcholine (CHRNG) et celui codant la tropomyosine 2 (TMP2). Le premier est nécessaire au bon développement et à la mise de la jonction neuromusculaire pendant la vie fœtale, le relais étant ensuite pris par la sous-unité epsilon du même récepteur. Le second (TPMP2) intervient dans la mise en place et le fonctionnement de l’appareil contractile. Le phénotype observé dans cette série tunisienne est conforme au tableau classique de syndrome d’Escobar. Seul le patient 4 ne présentait pas de petite taille, cette exception ayant toutefois déjà été rapportée dans la littérature. Cette étude de cas confirme l’hétérogénéité génétique du syndrome d’Escobar. La littérature fait état de 83 cas de syndrome d’Escobar dont 75 étaient en lien avec un variant pathologique du gène CHRNG. Sur les 26 mutations de CHRNG répertoriées dans les bases de données, deux sortent du lot : une mutation faux-sens (c.459dup ; (p.V154Sfs*24)) et une délétion (c.753_754del ; (p.Val253Alafs*44)), cette dernière étant retrouvée dans la présente cohorte. Les investigateurs font, à juste titre, l’hypothèse d’un effet fondateur au Maghreb au regard de la relative grande prévalence de cette affection dans cette partie de l’Afrique du Nord. L’intérêt de cette publication réside dans le nombre important de cas présentés, la qualité de la description clinique et les études génétiques suggérant un possible effet fondateur. Manquent néanmoins des précisions sur les données électrophysiologiques, notamment concernant la recherche d’un éventuel décrément, ce qui aurait constitué un argument en faveur d’un dysfonctionnement de la jonction neuromusculaire. Une biopsie musculaire, surtout pour les cas avec mutation du gène TMP2, aurait également été intéressante, l’idée étant d’y rechercher des bâtonnets.
Des phénotypes cliniques hétérogènes dans les neuropathies motrices pures associées aux mutations du gène HSPB1
Heterogeneous clinical phenotypes of dHMN caused by mutation in HSPB1 gene: a case series
Actualité commentée réalisée par Jon Andoni Urtizberea - Publiée le 01 février 2023
TKMR-N 2023 ; 2 (3) : A3
Shen X, Zhang J, Zhan F, Tian W, Jiang Q, Luan X, et al.
Biomolecules 2022 ; 12 : 1382-96.
AVIS D’EXPERT
La multiplicité des dénominations correspondant aux neuropathies motrices héréditaires illustre bien la relative confusion qui règne les concernant. Neuropathie motrice distale (dHMN), maladie de Charcot-Marie-Tooth spinale (CMT spinale), ou amyotrophie spinales distale (dSMA) correspondent en fait aux mêmes entités. Le concept recouvre, en théorie, les neuropathies motrices héréditaires à prédominance distale sans composante sensitive. Parmi elles, la distinction n’est pas non plus toujours aisée entre motoneuronopathies et les axonopathies. A cela s’ajoute le fait que plusieurs gènes sont connus pour donner tout à la fois une neuropathie motrice pure ou une neuropathie sensori-motrice de type CMT comme dans le cas présent. C’est le cas notamment pour les gènes HSPB1, GARS, IGHMPB2, TRPV4, DNM2 et DYNCH1H1. La frontière entre neuropathie distale motrice pure et CMT reste très ténue, comme illustré par cette observation. Par ailleurs, la frontière avec certaines formes de SLA a été également rapportée dans la littérature. Au vu des anomalies observées à la biopsie d’un nerf sensitif, ici chez deux patients, il serait licite de reclasser ces observations parmi les cas de CMT de type 2F, ce que les auteurs n’ont pas souhaité faire. Ils ont considéré qu’une atteinte sensitive généralement très discrète, et surtout paraclinique, peut être tolérée sans sortir du cadre des dHMN. Ils soulignent de plus que cette composante peut devenir apparente très longtemps après le début des symptômes moteurs. L’article permet incontestablement d’enrichir nos connaissances sur ce groupe de pathologies complexes et à la nosologie pas tout à fait stabilisée. Dans le cas précis des formes liées à des variants pathogènes de HSPB1, les auteurs font état de l’association de traits relativement originaux comme un début précoce dans la première famille, la présence d’un syndrome de jambes sans repos dans la deuxième, et la diffusion des anomalies électromyographiques au-delà des seuls membres inférieurs. Ceci pourrait toutefois n’être qu’une coïncidence et/ou relever de l’anecdote. Au niveau génétique, le tableau des 40 mutations de HSPB1 déjà rapportées s’enrichit désormais d’un nouveau variant (p.V97L). Les études fonctionnelles pour prouver sa pathogénicité sont de grande qualité et les résultats très convaincants.
Une réexpression inégale de la dystrophine après une excision exonique obtenue par CRISPR dans le modèle double knock-out dystrophine/utrophine de la dystrophie musculaire de Duchenne
Non-uniform dystrophin re-expression after CRISPR-mediated exon excision in the dystrophin/utrophin double-knockout mouse model of DMD
Actualité commentée réalisée par Jon Andoni Urtizberea - Publiée le 17 janvier 2023
TKMR-N 2023 ; 2 (2) : A2
Hanson B, Stenler S, Ahlskog N, Chwalenia K, Svrzikapa N, Coenen-Stass AML, et al.
Mol Ther Nucleic Acids 2022 ; 30 : 379-97.
AVIS D’EXPERT
Fruit de manipulations des gènes Utrn et Dmd chez la souris, le modèle double knock-out (dKO) de la DMD se rapproche beaucoup plus de la pathologie humaine que la souris mdx, sa congénère naturelle spontanément mutée pour le gène Dmd. L’utrophine, une protéine musculaire structurellement très proche (dite paralogue) de la dystrophine, est significativement surexprimée chez la souris mdx mais aussi chez l’homme atteint de DMD. Le modèle dKO permet donc de s’affranchir de la compensation induite par l’utrophine, d’où un phénotype plus sévère et une espérance de vie beaucoup plus courte. De ce point de vue, les travaux à visée thérapeutique présentés ici sont de grande valeur car collant plus à la réalité clinique. Ces mêmes travaux démontrent aussi, s’il en était besoin, que l’édition génomique est arrivée à maturité et qu’elle recèle un potentiel sans doute très grand en matière de traitements personnalisés de nombreuses maladies génétiques. L’article cité en référence vient toutefois tempérer l’enthousiasme légitimement suscité par les premières expérimentations du système d’édition génomique CRISPR-Cas9 dans la DMD. L’édition génomique se révèle certes un outil très puissant pour corriger toutes sortes d’anomalies génétiques mais ses limitations, dans le contexte de la DMD, comme dans d’autres pathologies, ne sont pas négligeables. En premier lieu, la restauration du phénotype est, dans le cas présent, imparfaite. La réexpression de la dystrophine se révèle non-uniforme au niveau de la membrane musculaire et ceci pourrait expliquer en partie pourquoi les souris traitées ne survivent pas plus longtemps que leurs homologues non-traitées. Plus préoccupants, peut-être, sont les dommages collatéraux observés : le système CRISPR-Cas9 agit en effet comme des ciseaux moléculaires. À ce titre, il est susceptible d’entrainer des accidents/incidents génomiques liés à des coupures et des réarrangements à d’autres endroits du génome que ceux ciblés par le système. Ceci concerne également l’intégration de tout ou partie du génome du vecteur viral supposément non-intégratif. Comme toujours, la prudence s’impose donc avec les nouvelles technologies de pointe. D’autres expérimentations seront nécessaires pour les valider avant un passage chez l’homme.
La chloroquine corrige le défaut de taille des lysosomes dans des cellules déficientes en FIG4 et diminue la neurodégénérescence observée chez les souris knock-out Fig4
Chloroquine corrects enlarged lysosomes in FIG4 null cells and reduces neurodegeneration in Fig4 null mice
Actualité commentée réalisée par Jon Andoni Urtizberea - Publiée le 02 janvier 2023
TKMR-N 2023 ; 2 (1) : A1
Lenk GM, Meisler MH.
Mol Gen & Metab 2022 ; 137 : 382-87.
AVIS D’EXPERT
Le gène FIG4 est, lorsqu’il est muté, à l’origine de plusieurs maladies phénotypiquement très éloignées : d’une part, le syndrome de Yunnis-Varon responsable d’une atteinte multisystémique et létale impliquant le système nerveux central (SNC) et les os, et la CMT autosomique récessive de type 4J, d’autre part. Les cellules déficientes en FIG4 ont comme particularité de présenter des altérations des lysosomes, la protéine codée chez l’homme par FIG4 intervenant dans la biosynthèse et le renouvellement du signal-lipide référencé PI(3,5)P2 situé dans la membrane du lysosome.
Dans les modèles cellulaire et murin de ces pathologies, les lysosomes sont de taille anormalement grande et leur pH est significativement abaissé. Ces deux biomarqueurs sont relativement faciles à mesurer et à reproduire. Les chercheurs ont également pu mesurer indirectement l’immunofluorescence d’un marqueur lysosomal pour juger de l’effet biologique du traitement.
Les résultats obtenus, tant dans les cellules déficientes en Fig4 que chez la souris transgénique, sont très probants. La chloroquine a indiscutablement un effet bénéfique in vitro et in vivo. Ceci permet d’envisager des développements cliniques, notamment dans cette forme de CMT dans laquelle aucune alternative thérapeutique n’existe.
Pour autant, ces effets semblent partiels comme en témoigne la non-modification des vitesses de conduction nerveuse. L’impact serait donc plus important sur le SNC et moindre en périphérique. De plus, comme le soulignent les auteurs, ces effets ont tendance à s’épuiser assez rapidement, les souris traitées ne dépassant pas l’âge de deux mois, par exemple.
Enfin, une certaine prudence s’impose concernant l’usage de la chloroquine en général. Celle-ci a déjà fait couler beaucoup d’encre pendant la pandémie de SARS-Cov-2. Il y a souvent un gap entre les données expérimentales, notamment au niveau cellulaire, et les applications médicales de ce produit. Par ailleurs, il est bien connu que son utilisation au long cours est susceptible d’entrainer une myopathie de type vacuolaire.
Le spectre clinique de la SMA-PME et la normalisation in vitro de son profil céramidique
The clinical spectrum of SMA-PME and in vitro normalization of its cellular ceramide profile
Actualité commentée réalisée par Jon Andoni URTIZBEREA - Publiée le 13 décembre 2022
TKMR-N 2022 ; 1 (7) : A15
Lee MM, McDowell GSV, De Vivo DC, Friedman D, Berkovic SF, Spanou M, et al.
Ann Clin Transl Neurol 2022. Online ahead of print.
AVIS D’EXPERT
La SMA-PME représente une forme très rare d’amyotrophie spinale non liée au chromosome 5q dont un peu plus d’une trentaine de cas a été rapportée à ce jour. Elle est due à des mutations du gène ASAH1 codant la céramidase acide, une enzyme lysosomale. Ce même gène est à l’origine de la maladie de Farber. Dans de très rares cas, le patient peut développer les deux maladies sans que l’on en comprenne bien les déterminants. Dans les deux présentations cliniques, et du fait du déficit enzymatique en céramidase acide, des résidus céramides toxiques s’accumulent dans plusieurs tissus dont le système nerveux central et, le cas échéant, la peau.
La SMA-PME doit être considérée comme une maladie neuromusculaire métabolique, d’origine lysosomale, et potentiellement accessible à un traitement substitutif. En étudiant les corrélations entre génotype et phénotype, les auteurs ont contribué à une meilleure appréciation de l’histoire naturelle de la maladie et à la constitution de groupes plus homogènes de malades, le tout en vue d’essais cliniques. Le fait que la grande majorité des patients ait un génotype identique ou presque identique devraient faciliter les choses.
La partie la plus originale de l’article concerne la preuve de concept du bénéfice d’une thérapie substitutive recombinante destinée à restaurer l’activité enzymatique céramidase acide dans des cellules appartenant à trois patients atteints de SMA-PME. D’autres auteurs avaient abouti au même résultat positif dans le modèle murin de la maladie de Farber, qu’il s’agisse d’une thérapie génique médiée par un lentivirus ou d’une enzymothérapie substitutive classique.
Au terme de ces travaux, deux grandes interrogations demeurent : premièrement, le très difficile passage de l’enzyme recombinante au niveau de la barrière hémato-méningée en limite les effets au niveau du système nerveux central. Or, ce tissu est la cible de choix dans la SMA-PME. L’autre question concerne la fenêtre de tir optimale pour commencer à traiter les patients. En d’autres termes, et comme noté dans d’autres stratégies thérapeutiques dans d’autres maladies de surcharge similaires, au-delà de quelle période d’accumulation des métabolites toxiques, l’enzymothérapie substitutive ou la thérapie génique n’arriveraient-elles pas après la bataille…
L’enzymothérapie substitutive in utero dans la forme infantile de la maladie de Pompe
In utero enzyme-replacement therapy for infantile-onset Pompe’s disease
Actualité commentée réalisée par Jon Andoni URTIZBEREA - Publiée le 12 décembre 2022
TKMR-N 2022 ; 1 (7) : A14
Cohen JL, Chakraborty P, Fung‑Kee‑Fung K, Schwab ME, Bali D, Young SP et al.
NEJM 2022. Epub ahead of print.
AVIS D’EXPERT
L’enzymothérapie substitutive à base de maltase acide recombinante constitue le traitement de base dans la maladie de Pompe. Il a déjà été démontré que l’ERT est d’autant plus efficace qu’elle est administrée très précocement. Le statut immunologique du patient peut être aussi un frein à la réussite du traitement. Les patients dits CRIM-négatifs produisent, dans des délais variables, des anticorps contre la maltase acide elle-même, celle-ci étant considérée comme une protéine étrangère lorsqu’elle est administrée sous forme de GAA recombinante.
Le plus souvent, le diagnostic d’IOPD est fait dans les premiers jours ou semaines de vie devant la constatation d’un ensemble de signes cliniques (hypotonie, détresse respiratoire, cardiomyopathie) et d’un effondrement de l’activité enzymatique GAA. Le diagnostic peut être fait aussi à un stade présymptomatique, le plus souvent dans les suites d’un dépistage néonatal que plusieurs États ont déjà mis en place à grand échelle (Taïwan, USA). Des traitements ultra-précoces ont déjà fait la preuve de leur efficacité, certains enfants ayant pu bénéficier de l’ERT dans les premiers jours suivant leur naissance, soit dans le cadre du dépistage soit à la suite de la découverte d’une cardiomyopathie in utero.
Le fait de traiter in utero un fœtus chez qui une IOPD a été diagnostiquée en prénatal constitue une avancée incontestable qu’il faut saluer. Le résultat obtenu, en tout cas avec 13 mois de recul, confirme le bien-fondé et l’innocuité de la méthode. Les modalités et le timing de cette intervention ne sont toutefois ni sans limitations ni sans risques. L’administration in utero de traitements de ce type est en effet réservé à des équipes très spécialisées. La morbi-mortalité liée au geste lui-même n’est pas négligeable, le risque étant bien évidemment celui d’une fausse-couche. L’ERT in utero doit être, selon nous, réservée à des cas extrêmes, notamment pour les couples qui ne souhaitent pas procéder à une interruption de grossesse suite à un diagnostic prénatal d’IOPD. Enfin, 13 mois de recul ne sont sans doute pas suffisants pour juger de l’efficacité de cette thérapie à long terme.
Caractérisation clinique et génétique d’une cohorte de patients brésiliens atteints de myopathie à bâtonnets
Nemaline myopathy in Brazilian patients: molecular and clinical characterization
Actualité commentée réalisée par J. Andoni Urtizberea - Publiée le 14 novembre 2022
TKMR-N 2022 ; 1 (6) : A13
Gurgel-Giannetti J, Santos Souza L, Yamamoto GL, Belisario M, Lazar M, Campos W, et al.
Int J Mol Sci 2022 ; 23 : 11995.
AVIS D’EXPERT
Bien que caractérisées, par définition, par un début précoce et une progressivité très lente, les myopathies congénitales constituent un groupe très hétérogène tant au niveau clinique que génétique. La myopathie à bâtonnets n’échappe pas à la règle comme le démontre cette étude conduite au Brésil chez 30 patients dûment sélectionnés3. Malgré la découverte récente de nombreux gènes morbides en rapport avec cette entité définie essentiellement au niveau histologique, il est encore très difficile d’établir des corrélations génotype-phénotype formelles.
Comme attendu, il ressort de cette étude que le gène de la nébuline (NEB) est de loin le gène le plus fréquemment mis en cause dans les myopathies à bâtonnets. La découverte d’un effet fondateur de ce gène est par ailleurs originale mais pas très étonnante dans le contexte d’un pays comme le Brésil où certaines populations ou régions peuvent être particulièrement endogames.
Le NGS s’affirme comme la technique de choix pour génotyper les patients atteints de myopathie congénitale en général et de myopathie à bâtonnets en particulier. Qu’il s’agisse de panels de gènes ciblés (couvrant l’ensemble des myopathies congénitales par exemple), de panels de gènes neuromusculaires, ou d’exomes entiers, le rendement diagnostique de ces méthodes est généralement très élevé. Les auteurs insistent, à juste titre, sur la nécessité de compléter ces études par la recherche de CNV, ce que de plus en plus d’algorithmes sont en mesure de faire, afin d’augmenter ce rendement. Cette approche double est particulièrement adaptée au cas du gène de la nébuline où il n’est pas rare que le deuxième variant pathologique manque à l’appel.
Au niveau clinique, outre la grande variation observée chez ces patients en termes de sévérité des symptômes, l’atteinte respiratoire, qui fait toute la gravité de ce type de myopathie une fois passée la période critique des premiers mois, doit faire l’objet d’une attention particulière. Une surveillance régulière clinique et paraclinique permet d’optimiser l’indication d’une ventilation assistée, le plus souvent non-invasive. On notera enfin l’intéressant travail d’inventaire des patterns d’imagerie musculaire en fonction des gènes en cause.
Une thérapie génique à base d’AAV prévient l’ossification hétérotopique observée dans la fibrodysplasie ossifiante progressive
Suppression of heterotopic ossification in fibrodysplasia ossificans progressiva using AAV gene delivery
Actualité commentée réalisée par J. Andoni Urtizberea - Publiée le 02 novembre 2022
TKMR-N 2022 ; 1 (5) : A12
Yang YS, Kim JM, Xie J, Chaugule S, Lin C, Ma H, et al.
Nat Comm Adv 2022 ; 13 : 6175-6195.
AVIS D’EXPERT
La FOP aussi appelée myosite ossifiante ou maladie de l’homme de pierre est une maladie orpheline touchant seulement quelques centaines de personnes à travers le monde. Transmise selon un mode autosomique dominant mais le plus souvent sporadique, elle associe des poussées d’ossification hétérotypique et des malformations osseuses, notamment un gros orteil de très petite taille très évocateur du diagnostic. La physiopathologie de la FOP n’est qu’imparfaitement connue mais tout laisse à penser qu’il s’agit d’une maladie du développement associée à un dérèglement de la réponse inflammatoire à des stimuli endogènes ou exogènes. 97 % des patients identifiés à ce jour sont porteurs de la même mutation du gène ACVR1 (p.R206H). Les biothérapies développées à ce jour dans la FOP sont avant tout pharmacologiques. Outre le traitement symptomatique des poussées d’ossification par les corticoïdes, l’arsenal thérapeutique s’est enrichi du palovarotene (un agoniste-gamma du récepteur à l’acide rétinoïque disposant d’une autorisation de mise sur le marché au Canada), et d’autres molécules en cours de développement clinique (anticorps anti-activine-A, rapamycine, inhibiteurs de l’ACVR1-kinase).
Les travaux cités en référence sont donc remarquables en ce sens qu’ils représentent une première et importante preuve de concept. Qui plus est, la technique de thérapie génique utilisée dans le cas présente est novatrice : elle combine à la fois le replacement du gène AVCR1 défectueux par un gène humain sain mais aussi l’introduction d’un micro-ARN artificiel permettant de rendre silencieux l’allèle pathologique.
Le virus associé à l’adénovirus (AAV) est bien connu des génothérapeutes et est largement utilisé en pathologie humaine, en particulier l’AAV-9. Ce dernier a fait, lors de ces travaux sur la FOP mais aussi dans l’amyotrophie spinale infantile en bien d’autres maladies génétiques, la preuve de sa grande efficacité dans la transfection des cellules mais également de sa bonne tolérance.
Cette étude marque une tournant dans la marche vers une application clinique de la thérapie génique dans la FOP. Elle est donc porteuse d’un grand espoir.
Fréquence des porteurs sains d’amyotrophie spinale en Arabie Saoudite
Spinal muscular atrophy carrier frequency in Saudi Arabia
Actualité commentée réalisée par J. Andoni URTIZBEREA - Publiée le 17 octobre 2022
TKMR-N 2022 ; 1 (4) : A11
Al Jumah M, Al Rajeh S, Eyaid W, Al-Jedai A, Al Mudhaiheem H, Al Sheri A, et al.
Mol Genet Genomic Med 2022 ; 00 : e2049.
AVIS D’EXPERT
La connaissance de l’épidémiologie d’une maladie génétique donnée, et a fortiori pour la SMA, revêt une importance capitale tant pour les professionnels que pour les autorités de santé. À l’heure où des médicaments très onéreux sont disponibles pour cette affection, les questions de stratégie pour mieux la combattre ou la prévenir sont régulièrement débattues.
Le caractère autosomique récessif de la transmission de la SMA la rend mécaniquement plus fréquente dans les zones de forte endogamie, quelle que soit la cause de cette dernière (isolement géographique, convictions religieuses, traditions culturelles, etc). L’Arabie Saoudite ne fait pas exception dans ce domaine.
Le travail conduit par Al-Jumah et ses collaborateurs est très intéressant et méritoire à la fois. Intéressant car il permet de disposer, pour la première fois, des chiffres précis d’incidence et de prévalence de la SMA dans cette région du monde. Méritoire car il n’est jamais très facile de réaliser ce genre d’études dans un pays aussi vaste et à l’intérieur duquel la population est inégalement répartie.
Le chiffre d’incidence (2,6 % d’hétérozygotes) obtenu est plus élevé que la norme (des études comparables font état de 1,5 % à 2 % dans d’autres populations notamment européennes), mais sans doute moins que prévu. On peut penser que ce chiffre est sous-estimé. Parmi les porteurs de deux copies du gène SMN1, par exemple, se cachent vraisemblablement quelques hétérozygotes dits 2-0 où les deux copies du gène sont situées sur le même allèle. Autre source potentielle d’erreur, un nombre important de cas (143) où le génotypage s’est avéré problématique.
Les auteurs incriminent, à juste titre, le degré élevé de consanguinité dans le royaume. Le chiffre de 27 % d’unions consanguines pourrait cependant paraître trompeur. Il ne s’agit en fait que de mariages entre cousins du premier degré. À cela s’ajoutent les liens de consanguinité plus ténus, mais réels, existant chez 19,3 % des individus, soit un total de 50 % de mariages endogames. Ces chiffres sont à rapprocher de ceux d’une étude pakistanaise1 dans laquelle 68 % des patients SMA étaient issus d’unions consanguines.
Ce travail va permettre aux Saoudiens de réfléchir à plusieurs scénarios de prévention de la SMA. Comme beaucoup d’autres pays de la région, ils devront choisir entre mettre en place un programme de dépistage néonatal et/ou un programme de dépistage des hétérozygotes, soit en population générale, soit en ciblant des couples en âge de procréer.
Les myopathies à agrégats tubulaires sont fréquemment d’origine génétique
Genetic defects are common in myopathies with tubular aggregates
Actualité commentée réalisée par J. Andoni URTIZBEREA - Publiée le 03 octobre 2022
TKMR-N 2022 ; 1 (3) : A10
Gang Q, Bettencourt C, Brady S, Holton JL, Healy EG, McConville J, et al.
Ann Clin Transl Neurol 2022 Jan ; 9 : 4-15.
AVIS D’EXPERT
Les myopathies à agrégats tubulaires, bien qu’ultra-rares, suscitent beaucoup d’intérêt de la part des chercheurs qui essayent de comprendre, outre le caractère inné ou acquis de ces affections, le lien qui pourrait exister entre l’apparition de ces structures anormales du réticulum sarcoplasmique et des manifestations cliniques généralement protéiformes et assez peu spécifiques. C’est le cas en particulier des spécialistes des syndromes myasthéniques congénitaux qui ont associé la présence de ces agrégats à certains gènes de la N-glycosylation (GDAP1, DPAGT1 et ALG2).
Les causes non génétiques de TAM ont déjà été rapportées dans la littérature comme l’exposition à certains toxiques, dont les médicaments (statines, corticoïdes) et l’alcool.
L’étude citée en référence fait ressortir que les causes génétiques représenteraient finalement un bon tiers des étiologies. Leurs présentations cliniques restent très variées. Aux signes myopathiques s’ajoutent des troubles oculaires lesquels peuvent constituer autant de signes d’appel.
Le rendement diagnostique de l’étude est, de ce point de vue, assez remarquable et tient au progrès permis par le séquençage à haut-débit. Les auteurs soulignent à juste titre que ce sont majoritairement des gènes défectifs dans la voie de signalisation du calcium (les gènes ORA1 et STIM1 en particulier) d’une part et dans la glycosylation de certaines protéines d’autre part (les gènes responsables de syndromes myasthéniques congénitaux). La question reste entière pour les deux autres tiers des patients étudiés. S’agit-il de gènes dont on ignore encore l’existence ? Ou d’authentiques formes acquises ? Cela reste à prouver.
Reste enfin qu’à une époque où le nombre de biopsies musculaires à visée diagnostique est en nette diminution, et ce au profit d’une approche NGS de première ligne, les TAM risquent de relever de l’anecdote. En revanche, la présence d’agrégats tubulaires pourra aider à la validation de variants dont la pathogénicité reste discutable.
Une étude d’innocuité et de tolérance de la corticothérapie dans les dystrophies musculaires de Becker et des ceintures
An open label exploratory clinical trial evaluating safety and tolerability of once-weekly prednisone in Becker and limb-girdle muscular dystrophy
Actualité commentée réalisée par J. Andoni URTIZBEREA - Publiée le 03 octobre 2022
TKMR-N 2022 ; 1 (3) : A9
Zelikovich AS, Joslin BC, Casey P, McNally EM, Ajroud-Driss S.
J Neuromuscul Dis 2022 ; 5 : 275-87
AVIS D’EXPERT
L’utilité de la corticothérapie dans la DMD repose sur un ensemble d’éléments de preuve provenant d’études ou d’essais cliniques1. Il n’en est pas de même pour les autres formes de dystrophie musculaire ou alors, de manière anecdotique et/ou contradictoire2. On ne retrouve par exemple qu’une étude allemande ayant prouvé de manière formelle que la corticothérapie était délétère dans les dysferlinopathies3.
L’étude ici commentée apporte un début de réponse tout en n’étant pas exempte de biais. La dose de corticoïdes reste, par exemple, très en-deçà de celle préconisée dans la DMD. On peut d’ailleurs douter de sa réelle efficacité au niveau biologique. La prise unique hebdomadaire est quant à elle un schéma d’administration très en vogue aux Etats-Unis et inspiré des pratiques observées en pédiatrie. On regrette également l’absence de données de suivi cardiologique alors que pour au moins trois des pathologies étudiées (BMD, déficit en FKRP et gamma-sarcoglycanopathie), le risque de développer une cardiomyopathie avec le temps est majeur.
On retiendra surtout de cet essai qu’effectivement, sur une durée d’observation non négligeable (deux années de suivi) et malgré l’évolutivité relativement lente des pathologies sélectionnées, la corticothérapie au long cours est non seulement bien tolérée mais est potentiellement efficace sur certains paramètres moteurs, et ce quel que soit le statut ambulatoire du patient. À tel point que 19 des 20 participants à l’étude ont décidé de la poursuivre une fois l’étude achevée. Pour autant, la grande hétérogénéité des formes de LGMD, les faibles effectifs de chaque sous-groupe, et surtout l’absence de groupe contrôle nuisent à la portée des résultats. Comme le soulignent les auteurs, une étude plus ambitieuse et plus centrée sur certaines formes de LGMD s’avère plus que jamais nécessaire à l’heure même où l’on dispose de plus en plus de données comparatives d’histoire naturelle dans ces pathologies.
Le bilan de l’ATU du risdiplam aux Etats-Unis
Expanded Access Program of risdiplam in the US
Actualité commentée réalisée par J. Andoni URTIZBEREA - Publiée le 03 octobre 2022
TKMR-N 2022 ; 1 (3) : A8
Kwon JM, Arya K, Kuntz N, Phan HC, Sieburg C, Swoboda KJ et al.
Ann Clin Transl Neurol 2022 ; 9 : 810-18.
AVIS D’EXPERT
Les données recueillies lors des phases d’ATU/EAP et, plus tard, en vie réelle, sont, pour les thérapies innovantes, toujours riches d’enseignements2. D’une part par la taille des populations de patients concernées et d’autre part du fait, le cas échéant, de périodes d’observation plus longues que lors des essais. L’étude américaine ici commentée ne déroge pas à la règle. Elle confirme que le risdiplam, administré per os à la différence des oligonucléotides antisens et de la thérapie génique médiée par l’AAV9, ne pose pas de problème majeur de toxicité. Elle confère ainsi au produit un incontestable avantage sélectif de ce point de vue. Même la toxicité rétinienne, un moment redouté en raison de données précliniques potentiellement inquiétantes, n’a pas été retrouvée lors de l’étude. Cette bonne tolérance est également illustrée par le fait que seuls trois patients sur les 155 ont arrêté le traitement du fait d’effets secondaires le plus souvent mineurs.
Les résultats de cette étude doivent cependant être relativisés et remis en perspective au vu du contexte américain de la SMA où l’accès aux traitements innovants est plus difficile qu’ailleurs et où les switchs d’une molécule à l’autre sont monnaie courante et peu discutés de manière collégiale. Ce type d’étude ne nous renseigne pas non plus quant au bénéfice réel du risdiplam au niveau clinique, ni sur les meilleures indications d’âge et de type de SMA.
Concernant la toxicité, enfin, on ne trouve pas trace du risque, désormais mieux documenté, de trouble de la spermatogénèse chez le garçon. Ce risque est d’ailleurs bien explicité aux individus concernés et à leurs familles, en tout cas en France.
Vers un élargissement de l’éventail phénotypique et génotypique de la neuropathie liée à HINT1
HINT1 neuropathy: Expanding the genotype and phenotype spectrum
Actualité commentée réalisée par J. Andoni URTIZBEREA - Publiée le 03 octobre 2022
TKMR-N 2022 ; 1 (3) : A7
Morel V, Campana-Salort E, Boyer A, Esselin F, Walther-Louvier U, Querin G, et al.
Clin Genet 2022 ; 1 : 1-12.
AVIS D’EXPERT
Les neuropathies à HINT1 sont de description récente (2012)1 et aucun cas n’avait été rapporté en France jusqu’à cette publication. D’après les données de la littérature, la prévalence de cette pathologie est plus élevée en Europe de l’Est et en Russie avec un effet fondateur très probable. Cette mutation fondatrice a d’ailleurs été retrouvée chez trois patients français. À noter que la cohorte comprend également le premier patient du continent africain (Algérie).
Le phénotype de cette maladie rare associe classiquement une neuropathie de type axonal et une myotonie d’intensité variable. Cette dernière peut être extrêmement subtile et n’être détectable que lors de l’examen électroneuromyographique systématique. Elle correspond à un état d’hyperexcitabilité d’origine nerveuse et peut se traduire par des myokymies. Mais elle peut être totalement absente comme décrit ici dans un quart de la cohorte française.
Des difficultés cognitives avaient déjà été pointées lors des premières descriptions de la maladie. Il peut s’agir de simples retards des apprentissages, notamment langagiers, ou de manifestations psychiatriques plus sévères pouvant aller jusqu’à un déficit intellectuel de sévérité variable ou à d’authentiques troubles de la personnalité. Le présent article des équipes françaises confirment cet aspect du phénotype lequel ne doit probablement rien au hasard.
Ces considérations cliniques sont à mettre en perspective avec les études fondamentales concernant le rôle de la protéine HINT1. Celle-ci a été impliquée chez l’animal dans le développement de troubles neuro-comportementaux superposables à la schizophrénie.
Les neuropathies liées à HINT1 revêtent donc un caractère original à plus d’un égard. Il est également probable que plus de patients seront diagnostiqués à l’avenir avec la diffusion du NGS au niveau des laboratoires de génétique moléculaire. On peut également faire le pari que la banalisation des études exome-entier au détriment des études plus fragmentaires sur panels de gènes CMT va aboutir à des phénotype liés à HINT1 mais sans composante neuromusculaire.
Mise au point et validation d’une échelle fonctionnelle du visage dans la myopathie facio-scapulo-humérale
Development and validation of the patient-reported “Facial Function Scale” for facioscapulohumeral muscular dystrophy
Actualité commentée réalisée par J. Andoni URTIZBEREA - Publiée le 03 octobre 2022
TKMR-N 2022 ; 1 (3) : A6
Karlien Mul K, Wijayanto F, Loonen TGJ, Groot P, Vincenten SCC, Knuijt S, et al.
Disabil Rehabil 2022 ; 16 : 1-6.
AVIS D’EXPERT
La Face Functional Score est la première échelle spécifiquement conçue pour la myopathie FSH par une équipe faisant autorité dans le domaine. Cet outil permet de mieux évaluer les déficiences des différents muscles du visage et surtout l’impact qui en résulte au niveau fonctionnel. Son élaboration a fait appel à des échanges fructueux avec les patients concernés, ceux-ci ayant fait part, en amont, de la pertinence des items du questionnaire initial. D’un point de vue biostatistique, les concepteurs ont également innové en utilisant une approche semi-automatisée de la méthode Rasch de validation des items, cette approche permettant un gain de temps non négligeable.
L’outil est toujours en cours de test pour étudier son évolution avec le temps. Il permettra peut-être de mettre en évidence et de quantifier une certaine évolutivité à ce niveau, très peu de données existant sur ce point. Il pourra être, le cas échéant, choisi comme outcome measure pertinent dans les essais thérapeutiques en cours de développement et/ou servir comme élément du suivi des patients en routine.
Si l’étude souffre certainement de la taille, relativement modeste, de l’échantillon et de quelques biais statistiques, elle constitue néanmoins une avancée significative. Elle n’existe pour l’instant qu’en langue hollandaise mais la validation de versions traduites dans d’autres langues est prévue. Reste enfin, et c’est un regret, que l’échelle ait été conçue et validée pour une population d’âge adulte (18 ans et plus). Or, les formes infantiles de FSH sont connues pour la sévérité et la précocité de l’atteinte faciale.
Dépistage néonatal de la dystrophie musculaire de Duchenne à Taïwan : résultats sur 50 000 nouveaux-nés testés
Duchenne muscular dystrophy newborn screening: the first 50,000 newborns screened in Taiwan
Actualité commentée réalisée par J. Andoni URTIZBEREA - Publiée le 03 octobre 2022
TKMR-N 2022 ; 1 (3) : A5
Yin‑Hsiu Chien, Ni‑Chung Lee, Wen‑Chin Weng, Li‑Chu Chen, Yu‑Hsuan Huang, Chao‑Szu Wu, et al.
Neurol Sci 2022 ; 43 : 4563-6.
AVIS D’EXPERT
Les premières expériences de dépistage néonatal dans la DMD réalisées en particulier à Chypre, Cardiff et Lyon à partir d’un dosage de CPK, se sont heurtées à un nombre important de faux-positifs1. Les CPK peuvent en effet être significativement augmentées pendant les premiers jours pour de multiples raisons (accouchement traumatique, injections intramusculaires, situations diverses et variées de stress, etc). De ce point de vue, le programme taïwanais ne déroge pas à la règle même si on peut s’interroger sur les seuils de positivité choisis. Son intérêt et son originalité résident dans le fait d’avoir mis en place, en complément, un test génétique de confirmation moléculaire permettant d’affirmer un diagnostic de certitude de DMD dans des délais très courts.
L’incidence de la maladie de Duchenne calculée à partir de cette étude laisse apparaitre une grande divergence avec l’incidence théorique communément admise pour la DMD (1 sur 3 500 naissances mâles). Il pourrait s’agir d’une spécificité locale, mais on peut en douter, ou simplement des limites technologiques du programme, le WES n’étant pas à même de détecter certains variants complexes du gène (mutations introniques, par exemple)2,3. Ceci requiert des investigations invasives qui ne peuvent être envisagées dans un cadre de dépistage de routine. Cette étude est également à remettre dans le contexte taïwanais. Ce pays a toujours été en pointe dans le dépistage à la naissance des maladies neuromusculaires, notamment pour la forme infantile de la maladie de Pompe (IOPD). Assez curieusement, les auteurs rapportent d’ailleurs un nombre plus important de cas d’IOPD dépistés que de cas de DMD.
Au-delà du dépistage lui-même, l’attitude thérapeutique à adopter face à ces nouveaux-nés par définition asymptomatiques reste sujette à discussion, qu’il s’agisse de la corticothérapie ou de thérapies innovantes comme l’ataluren. À partir de quel âge faut-il les introduire ? Comment gère-t-on la période intermédiaire ? Reste que le dépistage précoce de la maladie de Duchenne garde tout son intérêt pour le conseil génétique, permettant ainsi d’éviter la survenue de nouveaux drames dans les familles.
La cathepsine D présente dans le LCR de patients atteints de SMA traités par nusinersen pourrait être un biomarqueur
Cathepsin D as biomarker in CSF of nusinersen-treated patients with spinal muscular atrophy
Actualité commentée réalisée par J. Andoni URTIZBEREA - Publiée le 03 octobre 2022
TKMR-N 2022 ; 1 (3) : A4
Schorling DC, Kölbel H, Hentschel A, Pechmann A, Meyer N, Wirth B, et al.
Eur J Neurol 2022 ; 29 : 2084-96.
AVIS D’EXPERT
L’injection intrathécale d’oligonucléotides antisens de type nusinersen reste le traitement de référence dans l’amyotrophie spinale infantile. Son efficacité clinique a été prouvée à plusieurs reprises, lors des essais cliniques préliminaires mais aussi lors des études conduites en ouvert après l’obtention de l’autorisation de mise sur le marché. La tolérance du nusinersen est excellente si l’on met à part les syndromes post-PL et les difficultés d’administration liées aux anatomies complexes (rachis scoliotiques notamment). Pour autant, les cliniciens aimeraient bien disposer d’un marqueur biologique, aussi voire plus fiable que les échelles fonctionnelles déjà à leur disposition, afin de juger de l’efficacité du traitement et son éventuel épuisement dans le temps.
D’autres chercheurs avaient déjà souligné l’intérêt du dosage des neurofilaments dans le LCR dans les maladies neurodégénératives du motoneurone tout en soulignant la difficulté d’interprétation des résultats. La découverte des variations, ici à la baisse, de la cathepsine D, dans le LCR d’enfants traités pourrait donc être un outil très pertinent pour juger si le traitement administré continue d’être actif, au moins au niveau biologique. Les auteurs estiment que les variations conjointes de la cathepsine D et des chaines légères de neurofilaments seraient encore plus pertinentes, en tout cas dans la population pédiatrique.
L’étude souffre de la taille, relativement modeste, de l’échantillon de patients étudiés, surtout dans la phase préliminaire, et de l’absence de significativité statistique pour certains sous-groupes traités. Elle apporte néanmoins des éléments très intéressants que ses auteurs, et sans doute d’autres, voudront élargir à des échantillons de patients plus grands.
Le variant intronique c.1746-20C>G du gène CAPN3 est hypomorphe dans des formes récessives de calpaïnopathie (LGMD-R1)
CAPN3 c.1746-20C>G variant is hypomorphic for LGMD R1 calpain 3-related
Actualité commentée réalisée par J. Andoni URTIZBEREA - Publiée le 03 octobre 2022
TKMR-N 2022 ; 1 (3) : A3
Mroczek M, Inashkina I, Stavusis J, Zayakin P, Khrunin A, Micule I, et al.
Hum Mutat 2022 ; 43 : 1347-53.
AVIS D’EXPERT
Les calpaïnopathies primaires constituent le groupe le plus important des dystrophies musculaires des ceintures (LGMD pour Limb Girdle Muscular Dystrophy), en tout cas en Europe. L’existence de formes autosomiques dominantes (LGMD-D4) a eu beaucoup de mal à s’imposer malgré l’accumulation de publications allant en ce sens. L’article cité en référence est de nature à remettre en cause une partie de ces formes autosomiques dominantes dans la mesure où elle démontre qu’un variant peu ou pas recherché dans les études NGS standards, car situé dans une région intronique du gène CAPN3, pourrait avoir en fait un rôle pathogène et contribuer à l’expression de symptômes. D’où le terme d’ « hypomorphe » choisi par les auteurs pour qualifier ce variant. Il est probable que cet allèle intronique ne soit que le premier d’une longue liste à venir.
Ces données nouvelles et très convaincantes pourraient aider à résoudre un nombre non négligeable de cas sporadiques suspects de calpaïnopathie au niveau clinique mais chez qui un seul variant pathogène a été identifié en NGS standard ou en Sanger. Le variant c.1746-20C>G étant en amont du gène CAPN3 dans une région intronique, il échappe en effet au radar que constitue le séquençage qu’il s’agisse de Sanger ou de NGS (panels de gènes LGMD ou exome entier). Les études en génome entier permettront, à l’avenir, d’éviter cet écueil.
Au vu de la similitude du phénotype LGMD-R1 avec certaines formes de myopathie scapulo-huméro-humérale (FSH), il n’est pas exclu non plus que certaines de ces dernières formes négatives pour FSHD1 et FSHD2 soient en fait des LGMD-R1 avec un allèle CAPN3 hypomorphe.
Les auteurs ont étudié la fréquence allélique de ce variant et ont confirmé sa forte prévalence dans la région d’Europe de l’Est. Des études d’haplotypage pourraient aider à mieux localiser et dater l’origine de l’effet fondateur de cet allèle hypomorphe.
Une importante cohorte de patients canariens atteints de dystrophie musculaire oculopharyngée.
Clinical and genetic features of a large homogeneous cohort of oculopharyngeal muscular dystrophy patients from the Canary Islands.
Actualité commentée réalisée par J. Andoni URTIZBEREA - Publiée le 19 septembre 2022
TKMR-N 2022 ; 1 (2) : A2
Alonso-Pérez J, de León Hernández JC, Pérez-Pérez H, Mendoza-Grimón MD, Gutierrez-Martinez AJ, Hadjigeorgiou I, et al.
Eur J Neurol 2022 [Online ahead of print].
AVIS D’EXPERT
La DMOP est une pathologie neuromusculaire rare dont les caractéristiques sortent de l’ordinaire : l’atteinte musculaire y est très focale et son début d’apparition est très tardif. Sa physiopathologie l’est tout autant avec des expansions de triplets dont la taille est en partie corrélée à la gravité de l’atteinte clinique. L’étude des isolats génétiques a toujours été riche d’enseignements, et celui décrit dans les iles espagnoles des Canaries ne fait pas exception. On pense même que ce dernier a été à l’origine d’autres isolats comme celui en Uruguay, tel que rapporté par le regretté Mario Medici très récemment disparu [3], et un autre au Mexique. Les Canaries étaient en effet, aux 18e et 19e siècles, sur la route des colonies espagnoles d’Amérique Latine. Une écrasante majorité des patients canariens portent l’allèle (GCN)15 ce qui renforce l’idée d’un effort fondateur. De manière intéressante, les auteurs notent qu’à génotype identique, le phénotype des canariens concernés, serait globalement moins grave, preuve que d’autres facteurs, possiblement épigénétiques, jouent un rôle explicatif. De telles disparités entre génotype et phénotype avaient déjà été rapportées dans d’autres isolats pourtant génétiquement très homogènes comme celui du Québec. Les auteurs ont le mérite d’avoir réuni sur un territoire aussi restreint, une cohorte numériquement importante (113) si on la compare aux chiffres de prévalence communément admis en France (entre 350 et 400 patients ont été génotypés positivement pour la DMOP depuis la mise à disposition du test auprès des cliniciens) [4].
Le conseil génétique dans l’amyotrophie spinale infantile n’est pas exempt de pièges
Case report of pregnancy management and genetic evaluation after negative carrier screening for spinal muscular atrophy in an affected family.
Actualité commentée réalisée par J. Andoni URTIZBEREA - Publiée le 05 septembre 2022
TKMR-N 2022 ; 1 (1) : A1
Lucas HM, Sarumi MA.
Case Rep Womens Health 2021 ; 33 : e00377.
AVIS D’EXPERT
Dans la plupart des maladies génétiques autosomiques récessives, le conseil génétique est relativement aisé à condition de disposer de marqueurs biologiques univoques. Dans la SMA, les choses peuvent être beaucoup plus complexes comme l’illustre l’observation citée en référence. Le test génétique de base permet de compter le nombre de copies du gène SMN1 sans préjuger de sa répartition au sein des deux chromosomes 5q. Dans l’immense majorité des cas, les deux copies se trouvent de part et d’autre (on dit en trans) alors que chez 2 % des individus, les deux copies sont sur le même chromosome 5 (on dit en cis). Ce génotype particulier, dit aussi 2/0, correspond aux porteurs ou transmetteurs dits silencieux [2]. On peut suspecter cette situation très inhabituelle lorsque le phénotypage d’individus apparentés à un cas index de SMA fait apparaitre des personnes avec trois copies du gène SMN1.
Cette question du statut des hétérozygotes est d’autant plus prégnante que la prévalence des porteurs sains en population générale est très élevée dans la SMA (en moyenne autour de 1/50, encore plus dans les pays à forte endogamie) au point que certains pays, comme Israël, se sont lancés dans un dépistage des hétérozygotes à grande échelle. Ces dépistages ne peuvent, en l’état actuel des tests, détecter ce cas de figure complexe et sont de nature à faussement rassurer les individus souhaitant connaitre leur statut. Cette question se pose d’autant plus que l’on dispose désormais de thérapeutiques innovantes efficaces à condition d’être administrées très précocement.
Dans cette observation, le fœtus a eu la chance d’hériter de deux copies SMN1 de la mère et une copie SMN1 de son père porteur sain « traditionnel », et tout s’est bien terminé. Il en aurait été tout autrement si le père avait aussi été un transmetteur silencieux. Le fœtus aurait pu alors recevoir les deux allèles délétés de ses deux parents. Sa mère, comme l’ont prouvé des études complémentaires, était bien 2/0 et son père 1/1 au niveau de leur génotype SMN1 respectifs.
En France, et du fait des insuffisances et pièges de ce dépistage des hétérozygotes en population générale, la prudence est de mise, a fortiori s’il s’agit de mineurs. On peut miser sur les progrès technologiques des tests génétiques mais il faudra sans doute s’armer de patience.